Org. Synth. 2010, 87, 339
DOI: 10.15227/orgsyn.087.0339
ONE-POT SYNTHESIS OF 5H-INDAZOLO-[3,2-b]BENZO[d]-1,3-OXAZINE: TWO EFFICIENT PREPARATIVE METHODS
Submitted by Danielle M. Solano
1, Jeffrey D. Butler
1, Makhluf J. Haddadin
2, and Mark J. Kurth
1*.
Checked by Kyle D. Baucom and Margaret M. Faul.
1. Procedure
Caution: This procedure should be carried out in an efficient fume hood due to flammable gas evolution during the reaction.
A. 5H-Indazolo-[3,2-b]benzo[d]-1,3-oxazine. To a 500-mL round-bottomed flask equipped with a Teflon-coated magnetic stir bar (5 cm × 2 cm egg-shaped) is added 2-nitrobenzaldehyde (1) (4.08 g, 27.0 mmol, 1.0 equiv) and 2-aminobenzyl alcohol (2) (3.49 g, 28.3 mmol, 1.05 equiv) (Note 1). The solids are dissolved in methanol (70
mL), and glacial acetic acid (7.73 mL, 0.135 mol) is added to give a clear, yellow-orange solution (Note 2). 2-Picoline borane (3.50 g, 28.3 mmol, 1.05 equiv) (Note 3) is added in one portion, resulting in gas evolution and a mild exotherm (5 °C). The reaction flask is sealed with a septum, vented with a needle, and stirred for 3 h at room temperature at which point the reaction is darker orange in color. Reductive amination to (2-(2-nitrobenzylamino)phenyl)methanol is complete as indicated by the disappearance of 2-nitrobenzaldehyde by TLC analysis (Note 4). Additional methanol (200
mL) (Note 2) is added followed by a solution of potassium hydroxide (22.7 g, 0.405 mol) in water (30
mL) (Note 5). The septum is removed, the flask is fitted with a water-cooled reflux condenser, and the solution is heated, with stirring, to reflux (85 °C oil bath temperature, internal reaction temperature 72 °C) for 1.5 h to affect the bis-heterocyclization reaction (Note 6). The flask is removed from the oil bath, the solution is cooled to room temperature, and concentrated by rotary evaporation under reduced pressure (45-50 °C, 200-230 mmHg) to remove methanol, at which point a tan precipitate forms. The mixture is cooled to room temperature, and 1 M HCl (300
mL) is added (Note 7). The mixture is transferred to a 1-L separatory funnel, and ethyl acetate (200
mL) is added, which dissolves the precipitate. The organic layer is separated, and the aqueous layer is further extracted with ethyl acetate (2 × 100 mL). The combined organic extracts are washed with 1 M HCl (1 × 200 mL), 1 M K2CO3 (2 × 200 mL), and brine (1 × 200 mL). The organic extracts are dried over anhydrous sodium sulfate (40 g), filtered, and the solid residue is rinsed with ethyl acetate (2 × 50 mL). The combined extracts are concentrated by rotary evaporation under reduced pressure (45-50 °C, 200-230 mmHg). Purification by flash column chromatography on SiO2 (Note 8) affords 5.29-5.38 g of 5H-indazolo[3,2-b]benzo[d]-1,3-oxazine (3) as a white solid in 88.2-89.7% yield (Note 9).
Caution: This procedure should be carried out in an efficient fume hood. Appropriate precautions should be taken to avoid inhalation or direct contact with 2-nitrobenzyl bromide, a known lachrymator.
B. 5H-Indazolo-[3,2-b]benzo[d]-1,3-oxazine. To a 500-mL round-bottomed flask equipped with a Teflon-coated magnetic stir bar (5 cm × 2 cm egg-shaped) is added 2-nitrobenzyl bromide (4) (6.00 g, 27.8 mmol, 1.0 equiv) and 2-aminobenzyl alcohol (2) (4.11 g, 33.4 mmol, 1.2 equiv) (Note 10). The solids are dissolved in methanol (120
mL), and diisopropylethyl amine (DIEA, 9.5 mL, 55.6 mmol, 2.0 equiv) is added (Note 11). The reaction flask is fitted with a water-cooled reflux condenser, and the solution is heated to reflux (85 °C oil bath temperature, internal reaction temperature 73 °C) for 4 h. At this point, the displacement reaction is complete as indicated by the disappearance of 2-nitrobenzyl bromide by TLC analysis (Note 12). The solution is cooled to room temperature, then potassium hydroxide pellets (23.4 g, 0.417 mol) (Note 13) are added followed by water (12
mL). Upon addition, the solution turns orange in color and an exotherm (20 °C) is seen. The reaction vessel is again fitted with a water-cooled reflux condenser and heated to reflux to affect the bis-heterocyclization reaction (85 °C oil bath temperature, internal reaction temperature 72 °C). After stirring at this temperature for 1.5 h (Note 14), the flask is removed from the oil bath, the solution is cooled to room temperature, and concentrated by rotary evaporation under reduced pressure (45-50 °C, 200-230 mmHg) to remove methanol, during which time a heterogeneous emulsion is produced. The mixture is cooled in an ice/water bath, 1 M HCl (450
mL) is added (Note 15), and a white precipitate is observed. The mixture (room temperature) is transferred to a 1-L separatory funnel. Ethyl acetate is added (200
mL) to dissolve the precipitate. The organic layer is separated, and the aqueous layer is further extracted with ethyl acetate (2 × 100 mL). The combined organic extracts are washed with brine (1 × 200 mL), dried over anhydrous sodium sulfate (40 g), filtered, and the solid residue is rinsed with ethyl acetate (50
mL). The combined extracts are concentrated by rotary evaporation under reduced pressure (45-50 °C, 200-230 mmHg). Purification by flash column chromatography on SiO2 (Note 16) affords 4.87-5.10 g of 5H-indazolo[3,2-b]benzo[d]-1,3-oxazine (3) as a pale-yellow solid in 78.8-82.9% yield (Note 17).
2. Notes
1.
2-Nitrobenzaldehyde (98%) and 2-aminobenzyl alcohol (98%) were obtained from Sigma-Aldrich and used as received.
2.
Methanol from Sigma Aldrich (99%) and glacial acetic acid from JT Backer were obtained and used as received.
3.
2-Picoline borane (synonyms: borane-2-picoline complex, pic•BH
3) was obtained from Sigma Aldrich (95%) and used as received.
4.
Reaction progress can be monitored by TLC analysis (Silica gel 60 F
254 0.5 mm, Merck) using 30% ethyl acetate in hexane as the eluent with UV visualization (R
f 2-aminobenzyl alcohol = 0.10, R
f 2-nitrobenzaldehyde = 0.43, R
f (2-(2-nitrobenzylamino)phenyl)methanol = 0.26).
5.
Potassium hydroxide pellets were obtained from JT Baker.
6.
Reaction progress can be monitored by TLC analysis (Silica gel 60 F
254 0.5 mm, Merk) using 30% ethyl acetate in hexane as the eluent with UV visualization (R
f 2-aminobenzyl alcohol = 0.10, R
f (2-(2-nitrobenzylamino)phenyl)methanol = 0.26, R
f product = 0.45).
7.
The pH of the reaction was ~6 as measured with pH paper.
8.
Flash chromatography is performed using a 3.4 cm diameter column containing 120 g of SiO
2 (0.035-0.070, 60 Å, Acros Organics). The column is packed by adding 10% ethyl acetate in hexane to the dry silica and eluting several times (until no cracks or imperfections are visible). The crude material is prepared for loading by dissolving it in ethyl acetate, adding 10 g of Celite® 503 (J.T. Baker), and then concentrating to dryness by rotary evaporation (45-50 °C, 200-230 mmHg). The celite-impregnated material is dry-loaded onto the column. The system is eluted with 15% ethyl acetate in hexane and 50 mL fractions are collected. Fractions are examined by TLC (30% ethyl acetate in hexane as the eluent, product R
f = 0.45, UV visualization). All fractions containing the desired product are combined and concentrated by rotary evaporation under reduced pressure (45-50 °C, 200-230 mmHg), and then under high vacuum at 25 mmHg.
9.
Analytical data for 5
H-indazolo[3,2-
b]benzo[
d]-1,3-oxazine (
3): Mp 100-102 °C; IR (neat) ν
max 1631, 1530, 1503, 1460, 1157, 1095, 737 cm
-1;
1H NMR
pdf (400 MHz, DMSO-
d6) δ: 5.63 (s, 2 H), 6.93 (dd,
J= 8.2, 6.8 Hz, 1 H), 7.26 (ddd,
J= 8.9, 6.6, 1.0 Hz, 1 H), 7.39 (td,
J= 7.4, 1.0 Hz, 1 H), 7.48 (t,
J= 8.4 Hz, 2 H), 7.52 - 7.58 (m, 2 H), 7.88 (d,
J= 8.0 Hz, 1 H);
13C NMR
pdf (125 MHz, DMSO-
d6) δ: 67.9, 105.9, 115.1, 116.9, 119.1, 120.0, 121.8, 125.5, 127.0, 127.9, 129.6, 132.8, 143.4, 148.0; ESI MS
m/zcalcd. for C
14H
11N
2O ([M+H]
+) 223.1, found: 223.1. Purity was determined to be >99% by HPLC integration. HPLC specifications are as follows: electrospray (+) ionization, mass range 150 - 1500 Da, 20 V cone voltage, and Xterra® MS C
18 column (2.1 mm × 50 mm × 3.5 μm). The product is stable in the solid form under refrigeration for months; however minor decomposition has been observed when stored in solution at room temperature (i.e. TLC standards) over several days.
10.
2-Nitrobenzyl bromide was obtained from Alfa-Aesar (98+%) and used as received.
2-Aminobenzyl alcohol was obtained from Sigma-Aldrich (98%) and used as received.
11.
Methanol was obtained from Sigma Aldrich (99%) and used as received.
Diisopropylethyl amine (DIEA) was obtained from EMD (98%) and used as received.
12.
Reaction progress can be monitored by TLC analysis (Silica gel 60 F
254 0.5 mm, Merck) using 30% ethyl acetate in hexane as the eluent with UV visualization (R
f 2-aminobenzyl alcohol = 0.10, R
f (2-(2-nitrobenzylamino)phenyl)methanol = 0.26, R
f 2-nitrobenzyl bromide = 0.58).
13.
Potassium hydroxide pellets were obtained from JT Baker.
14.
Reaction progress can be monitored by TLC analysis (Silica gel 60 F
254 0.5 mm, Merck) using 30% ethyl acetate in hexane as the eluent with UV visualization (R
f 2-aminobenzyl alcohol = 0.10, R
f (2-(2-nitrobenzylamino)phenyl)methanol = 0.26, R
f product = 0.45).
15.
The pH of the reaction was ~1 measured with pH paper.
16.
Flash chromatography is performed using a 3.4 cm diameter column containing 120 g of SiO
2 (0.035-0.070, 60 Å, Acros Organics). The column is packed by adding 500 mL of 10% ethyl acetate in hexane to the dry silica and eluting several times (until no cracks or imperfections are visible). The crude material is prepared for loading by dissolving it in ethyl acetate, adding 10 g of Celite® 503 (J.T. Baker), and then concentrating to dryness by rotary evaporation (45-50 °C, 200-230 mmHg). The celite-impregnated material is dry-loaded onto the column. The system is eluted with 15% ethyl acetate in hexane and 50 mL fractions are collected. Fractions are examined by TLC (30% ethyl acetate in hexane as the eluent, product R
f = 0.45, UV visualization). All fractions containing the desired product are combined and concentrated by rotary evaporation under reduced pressure (45-50 °C, 200-230 mmHg), and then under high vacuum at 25 mmHg.
17.
Analytical data for 5
H-indazolo[3,2-
b]benzo[
d]-1,3-oxazine (
3): Mp 100-102 °C; IR (neat) ν
max 1631, 1530, 1503, 1460, 1157, 1095, 737 cm
-1;
1H NMR
pdf (400 MHz, DMSO-
d6) δ: 5.62 (s, 2 H), 6.93 (dd,
J= 8.2, 6.8 Hz, 1 H), 7.25 (ddd,
J= 8.9, 6.5, 0.9 Hz, 1 H), 7.38 (td,
J= 7.4, 1.0 Hz, 1 H), 7.48 (t,
J= 8.4 Hz, 2 H), 7.51 - 7.58 (m, 2 H), 7.88 (d,
J= 7.8 Hz, 1 H);
13C NMR
pdf (125 MHz, DMSO-
d6) δ: 67.9, 105.9, 115.1, 116.9, 119.2, 120.0, 121.8, 125.5, 127.0, 127.9, 129.6, 132.8, 143.4, 148.0; ESI MS
m/zcalcd. for C
14H
11N
2O ([M+H]
+) 223.1, found 223.1. Purity was determined to be >99% by HPLC integration. HPLC specifications are as follows: electrospray (+) ionization, mass range 150 - 1500 Da, 20 V cone voltage, and Xterra® MS C
18 column (2.1 mm × 50 mm × 3.5 μm). The product is stable in the solid form under refrigeration for months, however minor decomposition has been observed when stored in solution at room temperature (i.e. TLC standards) over several days.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The indazolobenzoxazine ring system (
3) is a heterocycle comprised of 2
H-indazole (
5) and dihydrobenzo-1,3-oxazine (
6) substructures (Fig. 1). Indazoles
3 and benzoxazines
4 are well represented in the chemical literature, and compounds containing these heterocycles display interesting biological activities.
5 Until our recent report,
6 the parent indazolobenzoxazine ring system (
3) and derivatives thereof were unknown and thus good candidates for discovery-based synthesis.
Figure 1. Heterocycles of interest: indazolobenzoxazine 3, 2H-indazole (5), dihydrobenzo-1,3-oxazine 6, and the 3-alkoxy-2H-indazole 7.
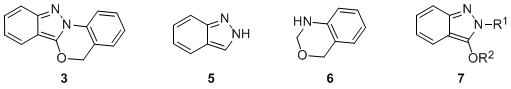
It has been demonstrated that 3-alkoxy-2
H-indazoles (
7) can be obtained from
o-nitrobenzylamines via an
N,N-bond-forming heterocyclization reaction mediated by potassium hydroxide in alcoholic solvent.
7 Further, we recently reported that an appropriately designed intramolecular variant of this reaction forms two fused heterocycles in a single step, providing an effective entry into the interesting indazolobenzoxazines and expanding the scope of this intriguing heterocyclization.
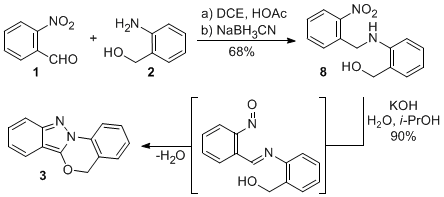
6 In this report, indazolobenzoxazine
3 was prepared stepwise by first performing a reductive amination of the imine resulting from the reaction of 2-nitrobenzaldehyde (
1) with 2-aminobenzyl alcohol (
2) to yield
o-nitrobenzylamine
8 (Scheme 1).
6 Bis-heterocyclization of
8 to
3, presumably via a nitroso imine intermediate, was achieved under basic conditions using aqueous KOH in isopropanol or methanol. It is of note that intramolecular cyclization (
8 →
3) is preferred to the potentially competing intermolecular possibility (e.g.,
8 →
7where R
2 is dependant on the solvent employed).
7 This stepwise approach, Scheme 1, delivered
3 in a 61% overall yield, demonstrating this to be a viable route for the preparation of 5
H-indazolo[3,2-
b]benzo[
d]-1,3-oxazines.
Scheme 1. Our initial stepwise route to the parent indazolobenzoxazine
3.
6
We then focused on optimization of our reported conditions to improve the efficiency and yield of this reaction. It was first determined that o-nitrobenzylamine 8 did not require purification; the crude material could be used in the subsequent bis-heterocyclization with no loss in yield. Further, it was established that switching the reductive amination solvent from 1,2-dicholoroethane (DCE) to methanol reduced reaction time without diminishing the yield. As the intramolecular bis-heterocyclization proceeded well in methanol, it seemed prudent to attempt both reactions in one-pot. Further modifications included a slight increase in the equivalents of 2-aminobenzyl alcohol (2) and addition of a potassium carbonate washing step to the workup. Finally, it was determined that the reaction time of the bis-heterocyclization could be significantly decreased by raising the reaction temperature to reflux. By integrating all of these modifications, indazolobenzoxazine 3 can be synthesized from 1 and 2 in one-pot, with a total reaction time of less than 6 h.
Sodium cyanoborohydride, the reducing agent of our initial approach, is highly toxic and residual cyanide is often detected in both the product and in generated waste.
8 We therefore proceeded to explore alternative reducing agents. Sodium borohydride successfully yielded indazolobenzoxazine
3, but in very low yield and purity. 2-Picoline borane (picBH
3) has been reported as an excellent alternative to sodium cyanoborohydride and has been used to perform reductive amination reactions in methanol.
8 To our delight, this reagent was perfectly compatible with our one-pot method. Using picBH
3, indazolobenzoxazine
3 is obtained in 88.2-89.7% yield, with a reaction time of less than 5 h (
Method A) and, more importantly, cyanide contamination is completely avoided.
Concurrently, we pursued an alternate route to o-nitrobenzylamine 8. Considering that 8 can also be prepared via N-alkylation, a one-pot protocol to synthesize indazolobenzoxazine 3 from 2-nitrobenzyl bromide (4) and 2-aminobenzyl alcohol (2) was explored. Though this nucleophilic displacement proceeds in methanol, reaction times are quite long (more than 24 h at room temperature). By heating to reflux, the reaction time was decreased to 4 h and the overall yield was increased. Subsequent addition of potassium hydroxide and further heating affords indazolobenzoxazine 3 in 78.8-82.9% yield (Method B). This method, although slightly lower in yield, significantly increases the scope of our methodology by extending the list of possible starting materials to include o-nitrobenzyl bromides. Further, properly modified benzyl alcohols (-OMs, -OTs, -OTf) are also potential starting points for the synthesis of indazolobenzoxazines.
In summary, we have developed two efficient, one-pot, scalable methods to prepare the 5H-indazolo-[3,2-b]benzo[d]-1,3-oxazine system, each from commercially available materials. Either method gives pure product in excellent yield (with Method A affording yields approaching 90%) and will allow access to a variety of novel indazolobenzoxazines.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2-Nitrobenzaldehyde; (552-89-6)
2-Aminobenzyl alcohol: Benzenemethanol, 2-amino-; (5344-90-1)
2-Picoline borane: Boron, trihydro(2-methylpyridine)-, (T-4)-; (3999-38-0)
5H-Indazolo-[3,2-b]benzo[d]-1,3-oxazine:5H-Indazolo[2,3-a][3,1]benzoxazine; (342-86-9)
2-Nitrobenzyl bromide: Benzene, 1-(bromomethyl)-2-nitro-; (3958-60-9)
Diisopropylethyl amine: 2-Propanamine, N-ethyl-N-(1-methylethyl)-; ( 7087-68-5)
 |
Mark J. Kurth received his B.A. in chemistry from the University of Northern Iowa in 1975 where he did undergraduate research with Professor James G. MacMillan. He obtained his Ph.D. from the University of Minnesota in the laboratory of Professor Thomas R. Hoye (1980) and subsequently did a one year postdoctoral study with Professor Wolfgang Oppolzer at the Université de Genève. He has been a faculty member in the Department of Chemistry at the University of California, Davis since 1981. His research interests focus on heterocyclic chemistry with emphasis on compounds of biological importance. |
 |
Danielle M. Solano received her B.S. in Biochemistry from the California Polytechnic State University, San Luis Obispo in 2004. She obtained her M.A. in Chemistry from Boston University in 2006, and is currently pursuing her Ph.D. at the University of California, Davis under the supervision of Professor Mark J. Kurth. Her research is focused on heterocyclic chemistry and includes methodology development, library synthesis, insecticidal applications, and the development of a4ß1 integrin antagonists towards the treatment of lymphoma. |
 |
Jeffrey D. Butler, originally from Oklahoma, received his B.S. degree in 2006 from California State University, Bakersfield where he worked in the laboratory of Professor Carl Kemnitz investigating the reactive intermediates of ozonolysis proposed by Rudolf Criegee. He is currently pursuing his Ph.D. at the University of California, Davis under the supervision of Professor Mark J. Kurth. His research is focused on the synthesis of novel heterocyclic libraries, asymmetric catalysis, and xenobiotics associated with primary bilary cirrocis (PBC). |
 |
Makhluf J. Haddadin received his B.Sc. and M.Sc. (Professor C.H. Issidorides) from the American University of Beirut, Lebanon; and his Ph.D. (Professor A. Hassner) from the University of Colorado, Boulder. He spent two years of post-doctoral work at Harvard University (Professor L.F. Fieser) and is currently Professor of Chemistry at the American University of Beirut. His research interests lie in synthetic heterocyclic chemistry. |
 |
Kyle D. Baucom obtained his B.S. degree in Chemistry from University of California at Santa Barbara in 2006 where he worked as an undergrad under the supervision of Jeffery Bode. In the fall of 2006 he began working at Amgen Inc. as a Process Chemist in the Chemical Process Research and Development group. He is currently a Senior Research Associate for Amgen. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved