SYNTHESIS OF 2-ARYL PYRIDINES BY PALLADIUM-CATALYZED DIRECT ARYLATION OF PYRIDINE N-OXIDES
Checked by Adnan Ganic and Andreas Pfaltz.
1. Procedure
A. 2-(4'-Methylphenyl)-pyridine N-oxide. A 1-L, three-necked round-bottomed flask is equipped with a magnetic stir bar (cylindrical, 4 × 1 cm), two glass stoppers and a reflux condenser with a gas bubbler on the top and connection to the argon line (Note 1). Pd(OAc)2 (0.560 g, 2.50 mmol, 5 mol%), PtBu3.HBF4 (0.870 g, 3.00 mmol, 6 mol%) (Notes 2, 3), potassium carbonate powder (K2CO3, 8.97 g, 65.0 mmol, 1.3 equiv) (Note 4), and pyridine N-oxide (19.0 g, 200 mmol, 4 equiv) (Note 5) are weighed in air and placed inside the flask. The whole setup is then evacuated (0.4 mmHg) and refilled with argon four times (Note 6).
A solution of 4-bromotoluene (8.55 g, 50 mmol, 1 equiv) (Note 7) in toluene (300 mL) (Note 8) is added under a steady flow of argon to the reaction mixture, and the glassware is rinsed with degassed toluene (30 mL) (Note 9). The obtained brown-orange suspension is immersed in the oil bath. Stirring (700 rpm) is commenced and the heating source is turned on (set to 125 °C). The mixture starts to reflux after approximately 45 min (Note 10).
Heating and stirring under a steady flow or argon is maintained for 16 h (overnight). The now black reaction mixture is allowed to cool to room temperature and transferred to 1-L round-bottomed flask. The reaction flask is rinsed with toluene (50 mL) (Note 11), and the solvent is removed under reduced pressure (40 °C, 40 mmHg) on a rotary evaporator. A saturated aqueous solution of NH4Cl (150 mL) and DCM (200 mL) are sequentially added to the dark slurry. The mixture is filtered through a Celite® pad (Note 12). The glassware is washed with water (50 mL) and DCM (2 × 50 mL). The resulting yellow biphasic solution is transferred into a 1-L separatory funnel and the layers are separated. The aqueous layer is washed with DCM (2 × 100 mL).The combined organic layers are dried over MgSO4 (4 g) (Note 13), filtered and washed with DCM (2 × 50 mL). The solvent is removed under reduced pressure (40 °C, 500 mmHg). The resulting yellow solid (Note 14) is then dissolved in eluent (20 mL) (Note 15), charged on a column (7 × 8.5 cm) of silica gel (Note 16) and eluted with DCM/acetone/MeOH 90:8:2 mixture (3.5 L). After the first 300 mL of eluent, fraction collection (20-mL fractions) is started. The desired product is obtained from fractions 18-120 as beige solid (6.51 g, 35.1 mmol, 70%) (Note 17).
The solid obtained after column chromatography is placed in a 250-mL round-bottomed flask equipped with a magnetic stir bar (egg shaped, 2 × 1 cm) and a reflux condenser. Heptane (40 mL) and toluene (20 mL) are added, and the suspension is heated to 115 °C (700 rpm). Toluene is added in 5 mL portions through the reflux condenser over 20-30 30 min until a clear solution is obtained (the total volume of toluene used is 40 mL). A gas bubbler is placed on top of the condenser and connected to the argon line. The heating source is turned off and the clear (yellow solution) is allowed to cool down to rt overnight (14 h) under argon, while stirring is maintained. The obtained suspension is cooled down to 0 °C with the help of an ice bath and stirred for 1 h. The resulting solid is collected by filtration using a 50-mL Büchner funnel under a stream of nitrogen and washed with ice-cold heptane (3 × 20 mL). The obtained solid is transferred to a 100-mL round-bottomed flask and dried overnight (>12 h) at 0.1 mbar to provide the title compound as a white solid (5.98 g, 32.3 mmol, 65%) (Notes 18, 19).
2. Notes
1.
Submitters setup: a 1-L pear-shaped flask is fitted with a Teflon sleeve and a cylindrical (4 × 1 cm) stir bar with a condenser.
2.
Palladium (II) acetate (46-1780) and phosphonium salt (15-6000) were purchased from
Strem and used without any purification.
3.
By using 10 mol% of the phosphonium salt the checkers did not observe any increase in yield (70%).
4.
Purchased from
Aldrich (347825) and used as is.
5.
Purchased from
Aldrich (131652), particles greater than 1 cm
3 are crushed using a mortar.
Pyridine N-oxide is a highly deliquescent solid,
3 and an undetermined amount of water is incorporated into the reaction mixture. The influence of water on the reaction was studied by the submitters. It is reported that the reaction occurs even when 5 equiv of water are added to the reaction mixture without affecting the yield.
4 An excess of this reagent is used to increase the yield of the reaction. The identical procedure can be performed with 1.5 equiv of
pyridine N-oxide resulting in yields ranging from 65-70% (checkers: 58%). During the extraction protocol the excess
N-oxide can be recovered from the aqueous phase by evaporation of water and chromatography of the residue with 10% MeOH in DCM.
6.
The submitters used a rubber septum on the top of the condenser, which allows connection to the vacuum line. The submitters added argon from a balloon. The checkers used argon from an argon line.
7.
Purchased from
Aldrich (B82200).
4-Bromotoluene is a low melting solid (mp 30 °C
). The submitters reported that it was stored in the freezer before use to facilitate its weighing as a solid. Alternatively, one could add it as a stock solution in toluene or as a liquid if heated. If it is added as a stock solution or a liquid, it should be added with the solvent. The checkers melted it using a water bath (40 °C) and weighed it as a liquid.
8.
The submitters used certified A.C.S. Grade toluene purchased from
Fisher Scientific (T324) and degassed it with argon (10 min) before use. The checkers used
J. T. Baker (Baker analyzed) toluene and degassed the solution by bubbling argon for 15 min prior to its use.
9.
Submitters reaction setup:
Toluene (330 mL) is added in 50 mL portions under a steady flow of argon. After addition of the first 50 mL, the reaction is immersed in the oil bath. Stirring (400 rpm) is commenced and the heating source is turned on (set to 125 °C). After the remaining toluene has been added the reaction is kept under an argon atmosphere and the stirring speed is raised to 700 rpm.
10.
The submitters report that the mixture starts to reflux after approximately 20 min and the color of the mixture pales to off-white after 30-40 min, which was not observed by the checkers. The reaction mixture remains heterogeneous, but slowly turns from brown-orange into a dark suspension.
11.
Not all material is dissolved in toluene, and therefore the reaction flask is rinsed with DCM and sat. aq.
NH4Cl solution, which is added to the dark slurry.
12.
Celite® (10 g) is weighed dry and packed using DCM (50 mL) in a 75-mL coarse fritted Büchner funnel. 20 g of sand is added to the top of the
Celite® layer.
13.
The submitters used 15 g of
MgSO4.
14.
In some cases a yellow oil is obtained which can be reduced to a yellow solid under high vacuum or longer time on rotary evaporator.
15.
The submitters used 20-35 mL of eluent for dissolving the material and report that sonication or heating with heat gun was necessary to fully dissolve the solid. The checkers dissolved the solid in 20 mL of eluent by stirring on a rotary evaporator without additional heating.
16.
The submitters used 175 g of
Silicycle (R10030B) silica gel on a 8.5 cm diameter column. 200 mL is collected in an Erlenmeyer flask followed by 15-mL fractions. The majority of the product was obtained from fractions 20-48. The product tailed off past fraction 55 but resulted only in a 2% increase in yield when collected. The checkers used silica gel (175 g) from
Fluka (89943) and a sand (50 g) layer on the top. The fractions between 121-160 contained product that was contaminated with unreacted starting material.
17.
The submitters report yields ranging from 79-83% (>97% purity by HPLC; contamination with <1% of tri-
tert-butylphosphine oxide; mp 129-131 °C). This material was usually carried through to further transformations, although analytically pure material could be obtained by recrystallization. Submitters HPLC conditions: Zorbax SB C18, RRHT, 1.8 micron (50 × 4.6 mm) 0.1%
H3PO4 in water / MeCN start 90:10, ramp to 50:50 over 8 min, ramp to 5:95 over 4 min. Total: 12 min, 2 mL/min, 35 °C, 215 nm, 5 uL. Retention times: Free base: 1.699 min,
N-oxide: 3.250 min. The checkers obtained yields in the 68-70% range (99% purity by HPLC; mp 141-143 °C). Checkers HPLC conditions: Reprosil 100 C18, 3 micron (125 × 3 mm), 0.1%
H3PO4 in water/MeCN start 80:20, ramp to 50:50 over 15 min, ramp to 15:85 over 15 min, total: 30 min, 0.5 mL/min, 35 °C, 254 nm, 1 mg/mL, 5 μL; retention time: 7.50 min.
18.
The checkers obtained yields after recrystallization in the 62-65% range (>99% purity by HPLC). Analytical data: mp 144-145 °C (lit.
5 mp 145-146 °C; TLC (
SiO2, DCM/Acetone/MeOH 90:8:2): R
f = 0.19;
1H NMR
pdf (400 MHz, CDCl
3) δ: 2.41 (s, 3 H), 7.14-7.23 (m, 1 H), 7.24-7.32 (m, 3 H), 7.41 (dd,
J = 7.8, 1.7 Hz, 1 H), 7.72 (d,
J = 6.6 Hz, 2 H), 8.32 (d,
J = 6.4 Hz, 1 H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 21.6, 124.3, 125.7, 127.3, 129.1, 129.3, 129.8, 139.9, 139.9, 140.7; IR (ATR)
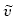
<: 3064, 3043, 1478, 1430, 1403, 1328, 1237, 1186, 1144, 1110, 1028, 1011, 948, 842, 817, 798, 735, 713, 694, 573, 526 cm
−1; MS (EI, 70 eV, 150 °C)
m/
z (%): 185 (M
+, 63), 184 (M-H
+, 100), 169 (10), 156 (13), 117 (25), 78 (12); Anal. calcd. for
C12H11NO: C, 77.81; H, 5.99; N, 7.56; found: C, 77.92; H, 6.22; N, 7.53.
19.
Submitters' recrystallization conditions:
N-oxide was suspended in 6 mL/g of
heptane in a three-necked flask fitted with a reflux condenser and a mechanical stirrer. The suspension is heated to 95 °C in an oil bath and toluene is added (in 1 mL/g portions) until all solids dissolve (typically ~6 mL/g). The heat source on the oil bath is turned off and mixture is allowed to cool in the oil bath overnight. In the morning, the suspension is immersed in an ice bath for 30 min and then filtered on a fritted Büchner funnel. The solid collected is then washed with 10 mL/g of heptane, and the solid is dried under a stream of N
2 and collected as a fluffy white solid which is >99% pure
(Note 17). Recovery for this procedure is typically from 88-95%.
20.
The Submitters used a mechanical stirrer.
21.
The submitters used ACS Grade THF with BHT over sieves from
A&C, which is used as is. The checkers use
J. T. Baker (Baker analyzed) material.
22.
Both submitters and checkers purchased zinc dust from
Aldrich (20,998-8) and used as received.
23.
The submitters stirred the reaction mixture for 40 min, and the reaction progress was checked by HPLC analysis. The checkers used TLC analysis (
SiO2, DCM/acetone/MeOH 90:8:2; R
f 0.87) to monitor the reaction progress.
24.
The submitters used
Solka floc®. They weighed Solka floc dry (15 g) and packed it using MTBE in a 60-mL coarse fritted Büchner funnel. The checkers used
Celite® (10 g), which was weighed dry and packed using MTBE in a 75-mL coarse fritted Büchner funnel, adding 10 g of sand to the top of the
Celite® layer.
25.
HPLC analysis shows >99% purity (for conditions, see Note 18; retention time 4.19 min). Analytical data:
1H NMR
pdf (400 MHz, CDCl
3) δ: 2.41 (s, 3 H), 7.20 (ddd,
J = 6.6, 4.8, 2.1 Hz, 1 H), 7.29 (d,
J = 7.9 Hz, 2 H), 7.64-7.78 (m, 2 H), 7.86-7.93 (m, 2 H), 8.65-8.71 (m, 1 H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 22.4, 120.4, 121.9, 126.9, 129.6, 136.7, 136.8, 139.1, 149.8, 157.6; IR (ATR)
: 3006, 2918, 1613, 1586, 1562, 1514, 1464, 1432, 1298, 1185, 1152, 1016, 829, 772, 741, 566, 529 cm
−1; MS (EI, 70 eV, rt)
m/
z (%): 169 (M
+, 100), 168 (49), 167 (16); Anal. calcd. for
C12H11N: C, 85.17; H, 6.55; N, 8.28, found: C, 85.13; H, 6.84; N, 8.15.
26.
In the submitters' procedure the product was not purified by distillation. It was stated that the isolated product was >98% pure (HPLC and exhibited identical spectral data as a commercial sample available from
Aldrich (98870). Storing of the obtained material is recommended under inert atmosphere, because the obtained product turns yellow under air at rt.
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The limited number of successful metal-catalyzed cross-coupling protocols using 2-metallapyridines has fueled the investigations of methods that do not require their use.
6 One strategy is to use direct arylation methodology, where a simple arene replaces the organometallic component.
7 In 2005, we reported that
pyridine N-oxide was a good substitute for
2-metallapyridine in cross-coupling reactions.
4,8 The
2-aryl pyridine N-oxide products can easily be converted to the corresponding 2-aryl pyridines under mild conditions and in high yield.
9 This protocol therefore allows for the rapid and easy synthesis of 2-aryl pyridines. More recently a full account describing optimized reaction conditions and reaction scope was reported.
10 The reaction tolerates a broad range of substitution patterns on both coupling partners. Substitution in the
ortho,
meta and
para position is well tolerated for alkyl and both electron-donating and electron-withdrawing substituents. The pyridyl moiety can be substituted at the 2-, 3- or 4-position allowing for a broad range of potential products. The reaction also proceeds with other azines such as isoquinoline and pyrazine. The procedure is convenient and makes use of cheap, readily available and air stable reagents. No precautions in the storage or the purification of the starting materials were taken. The 2-aryl pyridines obtained via this procedure are found in a number of biologically active compounds and are of value as organic synthesis building blocks. Furthermore, the
N-oxide moiety can serve as a useful handle in further elaboration of the pyridine core.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved