Org. Synth. 2011, 88, 162-167
DOI: 10.15227/orgsyn.088.0162
Discussion Addendum for: 5-ENDO-TRIG CYCLIZATION OF 1,1-DIFLUORO-1-ALKENES: SYNTHESIS OF 3-BUTYL-2-FLUORO-1-TOSYLINDOLE (1H-INDOLE, 3-BUTYL-2-FLUORO-1-[(4-METHYLPHENYL)SULFONYL]-)
Submitted by Junji Ichikawa.
*1Discussion
The precursors of 2-fluoroindoles, β,β-difluorostyrenes, are prepared by the palladium-catalyzed coupling reaction of 2,2-difluorovinylboron compounds with aryl iodides. The palladium-catalyzed reaction of dialkyl(2,2-difluorovinyl)boranes
2 (Scheme 1) with an aryl iodide resulted in the coupling of not only the difluorovinyl but also the alkyl moieties on the boron. The contamination by alkyl-coupling product was suppressed by using fluoride salt as a base,
2a,3 and eventually overcome by the selective oxidation of the alkyl-boron bonds leading to the boronates
3 prior to the coupling reaction as shown in the above scheme. Another way is a vinyl-selective transmetalation from boron to palladium via copper(I) species
6 (Scheme 1). On treatment of
in-situ generated
2 with aryl iodides in the presence of cuprous iodide and a palladium catalyst, disubstituted difluoroalkenes
7 are obtained in excellent yield (Table 1).
2b,2c,4bScheme 1
Alkenyl iodides and bromides,
5 and benzyl bromides
2c are also successfully employed in the cross-coupling reaction. The configuration of 1-alkenyl halides is completely preserved during the reaction. The coupling reaction with alkynyl iodides
6 and allyl bromides
2c proceeds without the palladium catalyst. These reactions provide important synthetic intermediates such as 1,1-difluoro-1,3- and -1,4-dienes and 1,1-difluoro-1,3-enyenes (Scheme 1, Table 1).
Table 1. Synthesis of Disubstituted 1,1-Difluoro-1-alkenes
The sequence of reactions: (i) the 1,2-migration via borate complexes and (ii) the coupling reaction via difluorovinylcoppers 6 provides a general synthetic method for unsymmetrically disubstituted 1,1-difluoro-1-alkenes 7 by the introduction of two different carbon substituents (R1 and R2) onto difluorovinylidene (CF2=C) unit in opposite polarities. That is, the carbon framework of difluoroalkenes can be constructed at will in this one-pot operation, where 2,2,2-trifluoroethyl p-toluenesulfonate (1) functions as a synthon of difluorovinylidene ambiphile (Scheme 2).
Scheme 2.
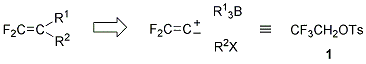
Thus, procedure A in the original article, providing
o-(1,1-difluorohex-1-en-2-yl)aniline, can be replaced with the following sequence (Scheme 3) on the same scale.
4a The solution of 2,2-difluorovinylborane, generated from
1 (15.3 g, 60 mmol), is treated with
hexamethylphosphoric triamide (HMPA, 20 mL), PPh
3 (1.26 g, 4.8 mmol), and Pd
2(dba)
3.CHCl
3 (1.24 g, 1.20 mmol). To the solution is added
N-butylmagnesio-
o-iodoaniline, which is generated
in situ from
o-iodoaniline (9.20 g, 42.0 mmol) and dibutylmagnesium (42 mL, 1.0 M in
heptane, 42 mmol) to avoid protonolysis of difluorovinylcopper
6. MeMgI can be also used for deprotonation of the amino group.
Copper(I) iodide (11.4 g, 60 mmol) is then added, and the reaction mixture is stirred at room temperature. After quenching the reaction with phosphate buffer, the mixture is treated with
hydrogen peroxide (100 mL, 30% in water) at 0 °C and then at room temperature for 1 h. The mixture is filtered through a pad of Celite, and organic materials are extracted with ethyl acetate. After removing HMPA by short column chromatography on silica gel, the crude product is distilled under reduced pressure to give
o-(1,1-difluorohex-1-en-2-yl)aniline (5.76 g, 65%). The coupling reaction of difluorovinylcoppers
6 can be also conducted with
o-iodo-
p-toluenesufonanilide without its deprotonation to give the indole precursors with an
N-tosyl group directly, albeit in slightly lower yield.
Scheme 3.
Moreover, difluorovinylcoppers
6 react as alkyl-substituted difluorovinyl anion with various electrophiles, such as acyl chlorides,
7 chlorodiphenylphosphine,
8 iodine,
9 NBS,
2c and (methylene)ammonium iodides,
2c which allows introduction of acyl, phosphine, iodine, bromine, and aminomethyl substituents to the difluorovinylic position. The directly-functionalized 1,1-difluoro-1-alkenes are readily supplied by this methodology.
While several methods for the activation of vinylboron compounds by transmetalation to copper have been reported,
10 all of them require a strong nucleophilic species, such as methylcopper, alkyllithium, or sodium methoxide, to induce borate-complex formation. In the above-mentioned transmetalation, the lithium fluoride formed
in situ acts as the nucleophile, which allows the selective activation of the vinyl group on boron under mild conditions (Scheme 1).
Furthermore, this type of activation with fluoride ion and copper(I) salt can also be applied to fluorine-free alkenylboranes.
2b The cross-coupling reaction of a
B-(1-alkenyl)-9-borabicyclo[3.3.1]nonane (BBN), generated
in situ via hydroboration of the corresponding 1-alkyene with 9-BBN, readily proceeds at room temperature within 1 h in the presence of cesium fluoride, cuprous iodide, and a palladium catalyst, while high temperature is normally required for efficient reaction rates in the Suzuki-Miyaura coupling (Scheme 4).
2b,2c,4b This is a useful activation method of alkenylboranes, which increases their reactivity as carbon nucleophiles.
Scheme 4.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
hydrogen peroxide (7722-84-1)
copper(I) iodide (7681-65-4)
heptane (142-82-5)
hexamethylphosphoric triamide (680-31-9)
o-iodoaniline (615-43-0)
 |
Junji Ichikawa was born in Tokyo in 1958. He received his B. Sc. and Dr. Sc. from the University of Tokyo under the supervision of Professor Teruaki Mukaiyama. He joined Kyushu University as an Assistant Professor in 1985. After working at Harvard University with Professor E. J. Corey (1989-1990), he moved to Kyushu Institute of Technology as a Lecturer. In 1999, he joined the University of Tokyo as an Associate Professor. He was then appointed Professor in Department of Chemistry, University of Tsukuba in 2007. His research interests lie in the area of synthetic methodology, specifically the development of novel reactions based on the properties of metals and fluorine.Biographical information can be typed here. Please limit the length of the biographical information to 100 words or fewer, if possible. Use Times New Roman, 12-point font with single spacing. The photographs should be submitted as separate JPEG files. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved