Checked by Alexander F.
G.
Goldberg and Brian M.
Stoltz.
1. Procedure
2. Notes
1.
Malonic acid (99%), phosphorous oxychloride (99%) and hexanes (mixture of isomers, ACS Grade, >98.5%) were was purchased from Sigma-Aldrich and used as received.
Aniline (99.9%) and toluene (ACS grade) were purchased from Fisher Scientific Company and used as received.
2.
The reflux condenser is fitted with an outlet that is connected with Tygon tubing to two 250 mL bubblers, the first empty, and the second filled with water to serve as the HCl trap.
3.
The gas flow should be monitored by constantly checking the bubbler during the aniline addition to prevent the development of negative pressure in the system.
4.
A creamy, yellow heterogeneous mixture is observed after completion of the aniline addition.
Some solids may aggregate near the surface and will slowly dissolve in the next stage of the reaction.
5.
Within the first 10 minutes of heating, vigorous gas evolution is observed as the mixture becomes homogeneous.
Within 20 min, the color of the solution transitions from yellow to red to dark brown/black.
Over approximately 90 min, the gas evolution subsides, and a gentle reflux is observed.
6.
If the solution is cooled for longer than 5 minutes, the reaction mixture may thicken to an extent that renders it difficult to transfer the entire contents of the flask to the quench beaker.
7.
This served to break up the largest fragments of material and to increase the surface area of the solid for efficient quenching in the following step.
8.
This workup procedure should fully hydrolyze all P-Cl bonds leaving sodium phosphate acid as the byproduct of POCl
3 quenching.
49.
This served to fully quench the suspension and to produce a finer crude solid with fewer chunks.
This facilitated transfer into the Soxhlet thimble and allowed for a more efficient extraction due to increased surface area of the solid.
10.
It may be necessary to filter the hexanes extract if red solid precipitates from the solution: The combined extracts were filtered through Celite® (21 g) in a coarse sintered glass funnel (46 mm diameter).
The filter cake was washed with hexanes (2 × 50 mL)
11.
The crude 2,4-dichloroquinoline (
1) exhibited the following: mp 62.0-64.2 (uncorrected); R
f = 0.42 (10% EtOAc/hexanes); IR (film) 3062, 1572, 1553 cm
−1;
1H NMR
pdf(400 500 MHz, CDCl
3) δ: 7.53 (s, 1 H), 7.67 (ddd,
J = 8.3, 6.9, 1.2 Hz, 1 H), 7.81 (ddd,
J = 8.5, 6.9, 1.4 Hz, 1 H), 8.05 (d,
J = 8.5 Hz, 1 H), 8.21 (dd,
J = 8.4, 1.4 Hz, 1 H);
13C NMR
pdf(125 MHz, CDCl
3) δ: 122.2, 124.4, 125.4, 128.1, 129.2, 131.48, 144.26, 148.3, 150.0; HRMS (APCI-ESI) calcd for C
9H
6Cl
2N [M+H]
+ 197.9872; found 197.9870.
This data matched the values given in the literature.
2 The checkers observed the development of a brown discoloration during prolonged storage at ambient temperature in air.
This does not affect its use in subsequent reactions.
12.
The submitters obtained yields between 33-36% using a smaller Soxhlet apparatus, which may have contributed to a greater extraction efficiency, with thimble dimensions: 33 mm × 94 mm (int.
diam.
x ext.
length).
13.
The crude material can be further purified by recrystallization.
The crude solid (5.0 g) was dispensed into a 50 mL Erlenmeyer flask and dissolved in 20 mL of a 3:1 mixture of hexanes and toluene with heating in a ~70 °C water bath.
The solution was allowed to slowly cool to room temperature
(Note 14) before it was placed in an ice-water bath.
After 2 hours, the mother liquor was decanted.
The recrystallization flask was kept in the ice-water bath while the crystals were rinsed with ice-cold hexanes (5 mL).
The hexanes were decanted and residual solvent was removed by vacuum (0.2 mmHg) to leave a yellow crystalline solid (2.99-3.17 g, 60-63% recovery,
(Note 15)).
14.
The submitters note that if a precipitate forms when the solution is cooled to room temperature, then the solution should be filtered before cooling in an ice-water bath.
15.
The melting point range of the recrystallized 2,4-dichloroquinoline (
1) was 64.1-65.0 °C (uncorrected).
The submitters note that Material material that was purified by column chromatography (0-4% EtOAc in hexanes) exhibited a melting point of 64.0-64.5 °C (uncorrected).
16.
The apparatus was flame-dried under reduced pressure (1-2 mmHg) and then maintained under a positive pressure of argon during the course of reaction.
17.
(±)-2,2'-Bis(diphenylphosphino)-1,1'-binaphthalene (97%) was purchased from Sigma-Aldrich and was used as received.
Sodium
tert-butoxide (97%) was purchased from Sigma-Aldrich, stored in a glovebox under a nitrogen atmosphere, and otherwise used as received.
Toluene (ACS grade) was purchased from Fisher and was dried by passage through a column of activated alumina as described by Grubbs.
5 The checkers used bis(dibenzylideneacetone)palladium as received from Sigma Aldrich and
(1R,2R)-(-)-1,2-diaminocyclohexane (99%) as received from Strem Chemicals.
The submitters prepared bis(dibenzylideneacetone)palladium according to the procedure of Rettig and Maitlis.
6 The submitters used (1
R,2
R)-(-)-1,2-diaminocyclohexane resolved from a
trans/
cis mixture (60/40) using the protocol of Jacobsen
7 with L-(+)-tartaric acid; the salt was then dissolved in 10 M NaOH and continuously extracted using benzene in a liquid/liquid extractor for 5 days.
This diamine is stored in a freezer.
18.
(1
R,2
R)-(-)-1,2-Diaminocyclohexane is a white solid but becomes a viscous oil as it warms to room temperature.
To avoid difficulties in transfer, the diamine is weighed directly into the reaction vessel without allowing the diamine to warm to room temperature.
19.
Sodium
tert-butoxide were dispensed into a over-dried vial in a glovebox under an nitrogen atmosphere.
This vial was sealed and removed from the glovebox and its contents were then quickly transferred into the round-bottomed flask.
20.
The reaction should be monitored very carefully for the first 10 minutes; the checkers consistently observed an exotherm within the first 5 minutes of heating.
When probed, the internal temperature gradually increased to 80 °C over 4 minutes of heating, then quickly rose to its boiling point within 30 seconds.
When the solution began to boil, the flask was removed from the oil bath and allowed to cool at ambient temperature until a precipitate formed (See
(Note 21)).
21.
After 5-10 minutes a solid formed and was manually dispersed after removal of the flask from the oil bath by vigorously swirling the flask by hand.
The flask was then returned to the oil bath.
22.
Thin layer chromatography (TLC) was performed using E. Merck silica gel 60 F254 precoated glass plates (0.25 mm).
UV light was used to visualize the product.
With 20% ethyl acetate in hexanes, the starting material has R
f = 0.40 and the product has R
f = 0.07.
Upon reaction completion, there may still be a relatively faint spot visible of the same R
f as the starting material.
23.
This material is of sufficient purity for use as a reactant to make compound
3.
The submitters note that it can be further purified by column chromatography (10% ethyl acetate in hexanes); the melting point of material purified by column chromatography is 236.0-237.0 °C.
The submitters note that the crude material exhibited some variation from batch to batch in terms of melting point;
a range of 235-245 °C was observed, but individual samples exhibited a routinely narrow (≤ 2 °C) melting point range.
H,
4ClQuin-BAM (
2) exhibited the following: mp 240.7-243.0; [α]
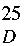
+568 (
c 1.02, DMSO); R
f = 0.31 (20% EtOAc in hexanes); IR (filmKBr) 3250, 2942, 1614 cm
−1;
1H NMR
pdf(500 MHz, DMSO-d
6) δ: 1.29-1.45 (m, 4 H), 1.71-1.79 (m, 2 H), 2.18-2.26 (m, 2 H), 4.05 (br s, 2 H), 6.84 (s, 2 H), 7.22-7.29 (m, 4 H), 7.53-7.62 (m, 4 H), 7.82 (d,
J = 8.0 Hz, 2 H);
13C NMR
pdf(125 MHz, DMSO-d
6) δ: 24.3, 31.8, 53.6, 112.6, 120.3, 122.1, 123.4, 126.0, 130.4, 140.2, 148.5, 156.5; HRMS (APCI-ESI) calcd for C
24H
23Cl
2N
4 [M+H]
+ 437.1300, ; found 437.1295.
The submitters have found H,
4ClQuin-BAM to be a bench stable solid: no decomposition of this compound has been observed after years of storage on the benchtop in a screw cap vial.
24.
Pyrrolidine (99%) was purchased from Alfa Aesar. α,α,α-trifluorotoluene (99+%) was purchased from Acros.
Dichloromethane (ACS grade) and acetone (ACS grade) were purchased from Sigma-Aldrich.
All reagents were used as received.
25.
Care should be taken to wash solids on the vial wall below the solvent level to prevent localized heating.
26.
The reaction was performed in a Biotage Initiator EXP US 355302 microwave reactor in a sealed vial, at the "Normal" absorption level.
This microwave system monitors the pressure and heat inside the sealed vial and shuts off for safety purposes when 20 bar or 250 °C is reached.
A two-stage heating protocol was found by the checkers to prevent an exotherm which exceeds the safety parameters of the microwave system; this was sometimes observed by the submitters when the reaction was heated immediately to 200 °C.
Using the described protocol, the checkers observed a temperature peak at approximately 185 °C during the initial heating stage.
During the second stage, the temperature was observed to peak at approximately 210 °C and the pressure at 11 bar; the temperature then stabilized at 200 °C, and the pressure at approximately 9.5 bar.
Different microwave reactors may have different safety parameters and vial headspaces:
caution is strongly recommended if different microwave instrumentation is employed.
27.
Confirmation of consumption of starting material can be made by TLC with UV visualization (10% methanol/0.5% acetic acid/89.5% dichloromethane, starting material R
f = 0.49 (checkers) (R
f = 0.79, reported by submitters); product R
f = 0.35).
28.
The contents of the microwave vial will congeal upon cooling, but the use of 20 mL of dichloromethane will sufficiently dissolve and transfer the mixture.
It might be necessary to stir the contents of the vial with the added dichloromethane for several minutes to get this congealed mixture to dissolve for transfer.
The use of an abnormally large flask (500 mL for less than 30 mL of solution) is necessary because this viscous mixture foams upon concentration by rotavap.
The extra headspace is needed to prevent the crude product from foaming out of the flask and into the rotavap bump trap.
29.
The submitters note that the material at this stage can be dried by subjecting the material to a drying pistol heated by refluxing toluene for 24-48 h and have found this dried material without further purification to give the same performance (stereoselection) as a catalyst as material further purified (column chromatography or recrystallization).
30.
The crude material might fully dissolve and crystallize in a matter of seconds upon addition of the solvent mixture at room temperature.
These crystals may not fully dissolve upon heating and swirling, but the recrystallization will still work when the solution is cooled.
31.
Vacuum was applied with a needle through a septum secured to the top of the flask.
The goal of this manipulation is merely to obtain a free-flowing solid.
32.
The submitters note that The the mother liquor and crystal washings were can be concentrated and subjected to the same recrystallization procedure to obtain an additional 800 mg of product.
The submitters note that during the determination of the melting point, the material gradually turns from white to slightly brown between 200 °C and the melting point.
Despite this appearance change, the melting point observed by the submitters is consistent at 243-245 °C.
The submitters have noticed some variation in the melting point range (241-246 °C), but individual samples exhibit a routinely narrow (≤ 2 °C) melting point.
PBAM (
3) exhibited the following: [α]
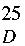
+398 (
c 1.2, CHCl
3); R
f = 0.35 (10% MeOH/CH
2Cl
2 with 0.5% acetic acid); IR (film) 3241, 3047, 2933, 2848, 1592, 1525 cm
−1;
1H NMR
pdf(500 MHz, CDCl
3) δ: 1.36-1.55 (m, 4 H), 1.75-1.89 (m, 10 H), 2.26-2.34 (m, 2 H), 3.05-3.16 (m, 4 H), 3.24-3.35 (m, 4 H), 4.10 (br m, 2 H), 5.28 (s, 2 H), 5.68 (br s, 2 H), 7.01 (dd,
J = 8.3, 6.8, 1.3 Hz, 2 H), 7.40 (ddd,
J = 8.2, 6.8, 1.4 Hz, 2 H), 7.65 (d,
J = 7.9 Hz, 2 H), 7.87 (dd,
J = 8.4, 1.3 Hz, 2 H);
13C NMR
pdf(125 MHz, CDCl
3) δ: 25.3, 25.7, 33.5, 51.7, 56.4, 93.0, 118.8, 119.3, 124.8, 126.5, 128.5, 150.0, 153.3, 158.5; HRMS (APCI-ESI) calc'd for C
32H
39N
6 [M+H]
+ 507.3236, ; found 507.3245;
Anal.
calcd for C
32H
38N
6: C, 75.85; H, 7.56; N, 16.59.
Found: C, 75.93; H, 7.44; N, 16.65.
The submitters note that PBAM purified by column chromatography has been stable upon storage in a screw cap vial on the benchtop for several years without sign of decomposition.
33.
The checkers observed a ~3% acetone impurity in the
1H NMR, along with a depressed melting point (135-163 °C); the submitters used a vacuum pump which reached 0.1 mmHg during the drying pistol stage.
The checkers employed this material in an enantioselective aza-Henry reaction of the Boc-protected imine of
o-tolualdehyde with nitroethane, as previously reported by the submitters, and obtained the same level of enantioselectivity as reported.
8
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
BisAMidine ('BAM') ligands were first described in 2004 and discovered to be effective catalysts - as their protic acid complexes (e.g.
3·HOTf, eq 1) - for the diastereo- and enantioselective aza-Henry reaction (eq 1).
The nitroalkane adducts are readily reduced, delivering
vic-diamines in a two step sequence and differentially protected at the amine nitrogens.
This class of catalysts, often referred to as chiral proton catalysts to emphasize the importance of the polar ionic hydrogen bond responsible for activation and orientation of the substrate, has since emerged as an increasingly general solution to reagent control of aza-Henry reactions.
The title compound (H,
4PyrrolidineQuin-BAM, 'PBAM')
8 was recently reported as a substantially more reactive and equally selective catalyst for the aza-Henry addition of nitroalkanes to aryl aldimines (eq 1).
9 This reagent, again as its triflic acid salt, expands the scope of the nitroalkane component, facilitating the stereocontrolled addition of a broad range of primary nitroalkanes and secondary nitroalkanes (e.g.
nitropropane).
This catalyst was discovered to be effective as a chiral Brønsted base in the alkylation of nitroalkanes using indolenine electrophiles (eq 2).
10 More recently, the addition of aryl nitromethane pronucleophiles were investigated, as these are notoriously difficult donors in the aza-Henry reaction.
11,12 In an interesting twist, the protonation state of the BAM ligand no longer affected the stereoselection in many cases.
13 Regardless, further modification of the fundamental PBAM backbone by installation of methoxy substituents (
4) furnished the addition products with high diastereo- and enantioselection (eq 3).
This development enabled the first enantioselective synthesis of (-)-Nutlin-3
13 (eq 3), the first potent small molecule inhibitor of p53/MDM2 discovered by Hoffmann-La Roche.
14The synthesis of PBAM described here is a significant improvement upon our previously reported procedure.8 The initial procedure relied upon similar reaction conditions but flash column chromatography was used to purify after each step.
The chromatographic purification for the final compound was particularly troublesome as large volumes of solvent (dichloromethane, methanol, and acetic acid) were required for the compound to fully elute from the column.
The products of these two reactions are now purified by straightforward washings, filtrations, and a recrystallization after the last step.
The scale of this procedure is nearly an order of magnitude greater than previously reported.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved