Org. Synth. 2012, 89, 394-403
DOI: 10.15227/orgsyn.089.0394
Discussion Addendum For:
Efficient Asymmetric Synthesis of N-tert-Butoxycarbonyl α-Amino Acids using 4-tert-Butoxycarbonyl-5,6-Diphenylmorpholin-2-one:
[R)-(N-tert-Butoxycarbonyl)allylglycine (4-Pentenoic acid, 2-[[(1,1-Dimethylethoxy)carbonyl]amino]-, (2R)-]
Submitted by Ryan E.
Looper
1 and Robert M.
Williams
*2
.
Discussion
The use of the diphenylmorpholinone template for the synthesis of optically enriched amino acid derivatives remains a highly selective, practical and predictable method.
3 While the chiral auxiliary is destroyed (by its eventual conversion to dibenzyl) and may not be ideally suited for synthetic applications on an industrial scale, it is a versatile tool for the preparation of research quantities of unusual or rare amino acid derivatives.
The sense of stereo-induction with this template is generally very high and very predictable with new substituents being added to the α-carbon anti- to the phenyl groups.
Further the reactivity of this template is highly flexible.
4 The α-carbon can react not only as an enolate, as described in the original
Organic Syntheses preparation, but can also serve as an electrophile (via the iminium ion), radical,
1,3-dipole (azomethine ylide)
5,6,7 or can be converted to and used as the α-phosphonate.
It should also be noted that both antipodes of the template are commercially available, which permits the synthesis of both enantiomers of a given target with equal efficiency.
Since the original report in
Organic Syntheses on the preparation of (
R)-(
N-tert-butoxycarbonyl)allylglycine a number of groups have used this allylglycine derivative for a variety of applications.
Most often it is incorporated as a metathesis precursor.
For example, Nolen and co-workers investigated the preparation of a more stable
C-glycosyl serine mimic (
5), to be used in investigations of the
O-glycosylation of serine residues which are a ubiquitous post-translational modification for controlling biological interactions (Fig.
1).
8 The allylglycine derivative
1, derived from the
Organic Syntheses preparation, served as a cross-metathesis partner with gluco-heptenitol.
Thus treatment of
1 and
2 with Grubb's 2
nd generation Ru-catalyst gave the cyclization precursor
3 in 70% yield.
Oxymercuration with the pendent alcohol generated the C-glycoside
4 in high yield and as a single diastereomer after reduction of the alkyl-mercurial species with Et
3B and NaBH
4.
The acetoxy derivative
5, poised for elaboration as a peptide fragment, can be obtained after hydrogenolysis and acetylation.
Figure 1. Application of cross methathesis to allylglycine derivatives.
Cross-metathesis between an allylglycine derivative and tetradecene, followed by hydrogenation, has also been used to create (
R)-aminoheptadecanoic acid (Fig.
2A).
This long chain amino acid has found use in the generation of truncated muraymicin analogues with increased cell permeability.
9 Ring closing metathesis (RCM) with two allylglycine units has also been reported. This technology permitted the synthesis of 14-membered macrocyclic peptides which were shown to be selective antagonists of the d and m opioid receptors (Fig.
2B).
10 Medium size rings can also be generated through RCM with allylglycine derivatives.
This has found use in the development of Smac (second mitochondria-derived activator of caspases) mimetics (Fig.
2C)
11 and in the development of matrix metalloproteinase (MMP) inhibitors inspired by the natural product cobactin T (Fig.
2D).
12Figure 2. Medicinally relevant small molecule derived from allylglycine methathesis reactions.
Both (R) and (S)-(N-tert-butoxycarbonyl)allylglycine are now commercially available, albeit generally 2-3 times more expensive than the oxazinone template itself.
As such, the oxazinone's inherent applicability might ultimately lie in the preparation of novel allylic amino acid derivatives.
Below we report on some recent examples in natural product synthesis, medicinal chemistry, and the generation of unnatural amino acid mimetics.
Funk and Greshock described the utility of their bromomethyl vinyl ketone equivalent (6-bromomethyl-4
H-1,3-dioxin,
6) for the synthesis of the naturally occurring (2
S,4
R)-4-hydroxypipecolic acid (
11) (Figure 3).
13 Alkylation of the oxazinone with
6 gave
7 as a single diastereomer.
Thermolysis of this intermediate triggered a retrocycloaddition to reveal the vinyl ketone
8, which underwent intramolecular Michael addition after removal of the
N-
tert-butoxycarbonyl group with TMSOTf.
Reduction of the ketone group in
9 with borane gave
10 as a single diastereomer.
Hydrogenolysis of the auxiliary provided
11 in good yield and high optical purity.
Figure 3. Allylic alkylation en route to (2S,4R)-4-hydroxypipecolic acid.
In 2004 we reported the use of the crotylglycine derivative
13 in the synthesis of cylindrospermopsin,
14 7-epicylindrospermopsin,
15 and 7-deoxycylindrospermopsin
16 (Figure 4).
Alkylation of the oxazinone with crotyl iodide gave
12 in 92% yield and as a single diastereomer.
Removal of the auxiliary then gave (
R)-(
N-
tert-butoxycarbonyl)crotylglycine in reasonable yield and with high optical purity (> 99 : 1 e.r.).
This was further processed to the
oxazinone-N-oxide 14, which underwent an intramolecular [3+2]-cycloaddition to give the tricyclic isoxazolidine
15, thereby setting the three contiguous stereocenters in the A ring of these natural products.
17 The key tricyclic intermediate
15 was then deployed in total syntheses of all three naturally occurring cylindrospermopsin alkaloids, through initial reductive cleavage of the N-O bond, differentiation of the two one-carbon arms and elaboration.
Figure 4. Allylic alkyation to access the cylindrospermopsin alkaloids.
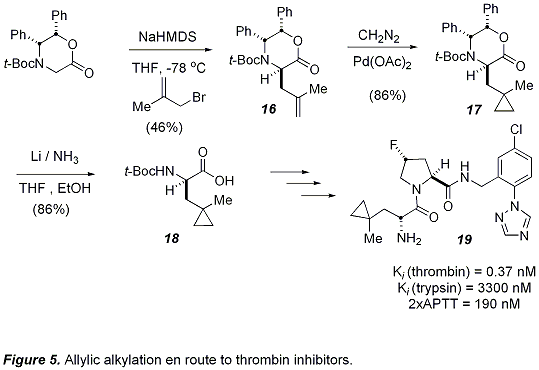
Researchers at Merck have utilized this methodology in the synthesis of a novel series of potent and selective thrombin inhibitors (Figure 5).
18 Alkylation of the oxazinone with
methallyl bromide gave the intermediate
16 in acceptable yield.
Cyclopropanation with diazomethane and Pd(OAc)
2 then gave the pendent cyclopropane in
17, which survived dissolving metal reduction of the auxiliary to give (
R)-(
N-
tert-butoxycarbonyl)-(methylcyclopropyl)propanoic acid (
18).
Incorporation of this fragment into their common fluoro-proline scaffold gave the thrombin inhibitor
19 (K
i = 0.37 nM), which was nearly 1,000 fold selective for thrombin over trypsin.
Figure 5. Allylic alkylation en route to thrombin inhibitors.
These types of reactions have been used in the preparation of other medicinally relevant compounds as shown in Figure 6.
Alkylation of the oxazinone with an allylic phthalimide bearing a vinyl fluoride gave
20, which has found use in the development of arginine mimetics as nitric oxide synthase inhibitors (e.g.
21) (Fig.
6A).
19 It should also be noted that the allylated oxazinone originally described in the
Organic Syntheses preparation can undergo a second round of alkylation to generate a quaternary α-carbon with good selectivity, again with the second electrophile approaching the enolate from the face opposite to the phenyl rings.
For example, alkylation with a pinacolatoborane reagent give the intermediate
22, which has found application in the synthesis of arginase inhibitors of the type
23 through oxidative cleavage of the allyl group and reductive amination (Fig.
6B).
20 A similar sequence can be carried out to alkylate the allylated oxazinone with BOMCl giving
24 (Fig.
6C).
Removal of the auxiliary and
benzyl ether provides (
S)-(
N-
tert-butoxycarbonyl)-(hydroxymethyl)-pentenoic acid (
25).
This intermediate has found utility in the preparation of pyrrolothiazoles as inhibitors of dipeptidyl peptidase IV (DPP-IV).
21Figure 6. Allylic alkylations to generate medicinally important small molecules.
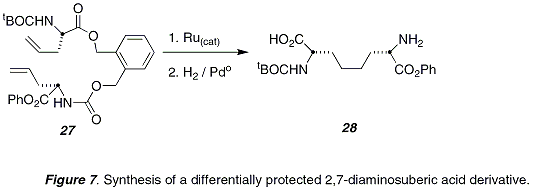
Another important use for allylglycine and its derivatives has been in the generation of conformationally constrained α-helical peptides.
An early report from our lab demonstrated that the tethered
bis-allylglycine substrate (
27) underwent olefin metathesis to give an E/Z-mixture of macrocyclic olefin products that upon hydrogenation provided the differentially protected 2,7-diaminosuberic acid derivative
28 (Figure 7).
22Figure 7. Synthesis of a differentially protected 2,7-diaminosuberic acid derivative.
The conceptually related intramolecular metathesis of allylglycine-containing peptides, has been pioneered by Verdine to make conformationally-constrained α-helical peptides which have been named "stapled peptides".
23 As shown in Figure 8A, the oxazinone template was used to synthesize both enantiomeric series of α-methyl-α-alkenyl amino acids.
The α,α-disubstitution increases helicity, while a variation in tether length allowed the examination of chemical yield for the metathesis reaction.
These amino acids were incorporated into model peptides at the i and i+4 positions.
Metathesis between the two alkene termini successfully "stapled" the peptides together.
The shortest metathesis reaction tolerated was that between the allylglycine derivative (
S-11) and hexenyl-substituted amino acid (
S-14) as shown in Figure 8B.
The opposite
R,R enantiomeric series was also successful while the
R,S series failed to metathesize.
An increase in tether length increased metathesis efficiency leading to reactions with >98% conversion.
Depending on the length of the tether, these peptides possessed greater helicity and resistance to proteolysis.
The original article detailing the preparation of (
R)-(
N-
tert-butoxycarbonyl)allylglycine provides a useful route to prepare this commonly used and now commercially available amino acid for the synthesis of more complex unnatural amino acids and elaborated peptides.
Perhaps more impressive is the utility of the oxazinone to prepare more ornate allylic-amino acid derivatives for use in natural products and medicinal chemistry.
Finally, the ease of preparation of the oxazinones
24 (also commercially available) allows for the synthesis of stable isotopomers of allylglycine, which can be introduced into the oxazinone template
25 via bromoacetate or via the allyl iodide fragment.
The incorporation of deuterium,
13C, and
15N into this system can thus be accomplished in several reasonably economical ways and can be of use in a variety of applications.
Figure 8. Synthesis of α,α-disubstituted amino acids and their incorporation into stapled peptides.
Adapted with permission from J.
Am.
Chem.
Soc. 2000, 122, 5891-5892, Copyright (2000) American Chemical Society.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
benzyl ether (103-50-4)
methallyl bromide (1458-98-6)
 |
Robert M.
Williams was born in New York in 1953 and attended Syracuse University where he received the B.A.
degree in Chemistry in 1975.
He obtained the Ph.D.
degree in 1979 at MIT (W.
H.
Rastetter) and was a post-doctoral fellow at Harvard (1979-1980; R.
B.
Woodward/Yoshito Kishi).
He joined Colorado State University in 1980 and was named a University Distinguished Professor in 2002.
His interdisciplinary research program (over 280 publications) at the chemistry-biology interface is focused on the total synthesis of biomedically significant natural products, biosynthesis of secondary metabolites, studies on antitumor drug-DNA interactions, HDAC inhibitors, amino acids and peptides.
|
 |
Ryan E.
Looper was born in Banbury, England in 1976.
He came to the U.S.
to attend Western Washington University where he earned a B.S.
degree in Chemistry in 1998 and an M.S.
degree in 1999 under the guidance of J.
R.
Vyvyan.
He obtained his Ph.D.
degree in 2004 at Colorado State University in the laboratories of Prof.
R.M.
Williams.
After a NIH post-doctoral fellowship at Harvard University with Prof.
S.L.
Schreiber, he began his independent career at the University of Utah in 2007.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved