Nugent's morpholinoisoborneol (MIB) is an excellent ligand for the catalytic asymmetric addition of alkyl, vinyl, and aryl groups to aldehydes in the presence of organozinc reagents to furnish a variety of secondary alcohols with high ee.
3,4,5 The major developments have been the significant broadening of the substrate scope and the development of several one-pot methods that streamline the synthesis of synthetically valuable and versatile molecules such as epoxy alcohols, allylic epoxy alcohols, cyclopropyl alcohols, halocyclopropyl alcohols, pyranones and 1,2,4-trioxanes with high enantio-, diastereo-, and chemoselectivity.
6,7,8 In this update, we will summarize many of the advances.
A detailed synthetic procedure for the synthesis of (-)-MIB has been published in
Organic Syntheses.
9 Starting from either (
R)- or (
S)-camphor, gram quantities of either enantiomer of MIB can be synthesized in three steps and with only a single purification step. At the time of this report, only (-)-MIB is commercially available.
Catalytic Asymmetric Arylation of Aldehydes
Two routes for the catalytic asymmetric arylation of aldehydes to generate highly enantioenriched diarylmethanols and benzylic alcohols were reported (Figure 1).
10 In the first route, diarylzinc intermediates were generated in situ by metalation of unfunctionalized aryl bromides with
n-BuLi followed by transmetalation with ZnCl
2 (Figure 1A). The LiCl formed in the process catalyzes a rapid racemic background reaction. The Lewis acidic LiCl was selectively suppressed by addition of tetraethylethylenediamine (TEEDA). Subsequent addition of catalytic amounts of (-)-MIB and aldehyde to the preformed diarylzinc reagent gave addition products with high enantioselectivities (80-92%) and yields (78-99%). In the second route, mixed alkyl aryl zinc reagents were generated by metalation of aryl bromides with
n-BuLi followed by addition to ZnCl
2 and subsequent addition of a second equivalent of
n-BuLi. The mixed organozinc species was used in situ in the asymmetric addition reaction after addition of 0.8 equivalents of TEEDA (Figure 1B). Higher enantioselectivities (up to 97% ee) were achieved via this route. This methodology was extended to the synthesis of aryl/heteroaryl- and diheteroarylmethanols with high levels of enantioselectivity.
11Figure 1. Catalytic asymmetric aryl additions to aldehydes with (A) Ar2Zn and (B) ArZnBu from aryl bromides.
Charette and coworkers introduced a complementary catalytic asymmetric arylation method by developing a salt-free preparation of diorganozinc reagents using Zn(OMe)
2 and alkyl/aryl Grignard reagents (Figure 2).
12 The insoluble salts Mg(OMe)
2 and/or NaBr salts were removed by centrifugation or filtration to afford the salt-free diorganozinc reagents. Two examples highlighting the efficiency of this chemistry are illustrated in Figure 2. Ishihara and coworkers adopted Charette's method to synthesize salt-free
i-Pr
2Zn and subsequently added it to aldehydes in the presence of 10 mol % (-)-MIB with up to 94% ee.
13,14Figure 2. Charette's catalytic asymmetric (A) alkyl and (B) aryl additions.
2. Synthesis of Chiral Acyclic Epoxy Alcohols
In an effort to streamline the enantio- and diastereoselective synthesis of valuable small molecules,
6 the (-)-MIB-based organozinc catalyzed carbonyl addition step was merged with several other transformations such as epoxidation, cyclopropanation, halocyclopropanation, and various rearrangement reactions. The first tandem reaction developed was for the synthesis of chiral epoxy alcohols.
Figure 3. Three one-pot synthesis of epoxy alcohols.
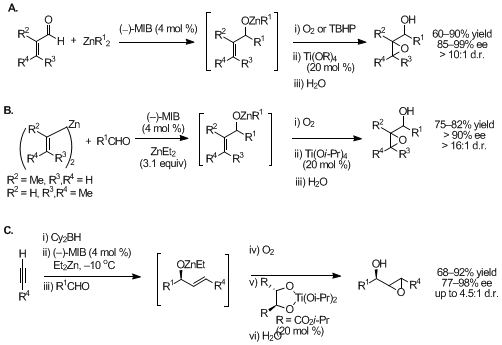
Two complementary methods were developed for one-pot synthesis of highly enantio- and diastereoenriched epoxy alcohols with up to three contiguous stereocenters.
15,16,17 The first route involved highly enantioselective alkyl additions to α,β-unsaturated aldehydes followed by titanium-catalyzed diastereoselective epoxidation with either dioxygen or TBHP (Figure 3A). The second route involves highly enantioselective divinylzinc additions to aliphatic or aromatic aldehydes (Figure 3B). The vinylzinc reagents can be either isolated and purified prior to addition to aldehydes,
18,19 or formed in situ by hydroboration of a terminal alkyne followed by transmetalation to zinc (Figure 3C).
20 The latter method provided access to a synthetically challenging class of secondary
trans-disubstituted epoxy alcohols with up to 4.5 : 1 dr. Excellent enantioselectivities were obtained with a wide range of aldehyde substitution patterns, except unbranched aldehydes, which undergo additions with up to 85% ee. This alkoxide-based titanium epoxidation catalyst is unique in that the
same catalyst demonstrated high diastereoselectivity with allylic alkoxides exhibiting either A
1,2 or A
1,3 allylic strain in one of the diastereomeric epoxidation transition states.
Synthesis of Chiral Allylic Epoxy Alcohols
Asymmetric vinylation of α,β-unsaturated aldehydes in the presence of catalytic amounts of MIB generated unsymmetrical bis(allylic) alkoxide intermediates. In situ alkoxide directed epoxidation afforded densely functionalized allylic epoxy alcohols in good yields and excellent chemo-, diastereo- and enantioselectivity (Figure 4).
16 The vinylzinc reagents may be either prepared and purified (Figure 4A) or generated in situ using Oppolzer's procedure.
20Figure 4. Synthesis of allylic epoxy alcohols using (A) purified vinylzinc reagents, or (B) in situ generated vinylzinc reagents.
The latter procedure afforded only (
E)-disubstituted vinylzinc reagents whereas the former allowed for more substituted vinylzinc reagents. The enal must bear non-hydrogen substituents in the R
2 or R
3 position so that either A
1,2 or A
1,3 strain is present in one of the diastereomeric epoxidation transition states. The unsymmetrical bis(allylic) alkoxide then underwent a highly chemoselective directed epoxidation of the more electron-rich double bond while minimizing A
1,2 or A
1,3 strain.
Catalytic Asymmetric (Z)-Vinylation of Aldehydes
The vinylation procedures above provide (
E)-allylic alcohols. Walsh and coworkers developed catalytic asymmetric (
Z)-vinylation of aldehydes with (
Z)-vinylzinc reagents via a novel 1,2-metalate rearrangement/transmetalation sequence.
21,22 Thus, hydroboration of 1-halo-1-alkynes followed by reaction with
tert-butyllithium,
23 transmetalation of the resulting (
Z)-vinylboranes to zinc and addition to prochiral aldehydes in the presence of (-)-MIB and the diamine inhibitor TEEDA furnished (
Z)-disubstituted allylic alcohols with high enantioselectivity and without contamination by (
E)-allylic alcohols (Scheme 1). This transformation is analogous to a net
trans hydroboration of the corresponding alkynes. Aliphatic aldehydes with α-branching gave products of high enantiopurity whereas β-branched aldehydes gave lower enantioselectivities.
Scheme 1. Catalytic asymmetric synthesis of (Z)-allylic alcohols.
4.1 Tandem Catalytic Asymmetric (Z)-Vinylation of Aldehydes/Diastereoselective Epoxidation and Cyclopropanation
The generation and addition of (
Z)-vinylzinc reagents to aldehydes were then applied to two one-pot tandem reactions. In the first tandem reaction, the resulting allylic alkoxides were treated with Et
2Zn, TBHP, and Ti(O
i-Pr)
4 to perform the diastereoselective and/or chemoselective epoxidation to synthesize epoxy alcohols and (
Z)-allylic epoxy alcohols (Figure 5A).
21 In the second tandem reaction, the allylic alkoxides were subjected to 5 equiv each of CF
3CH
2OH, Et
2Zn, and CH
2I
2 to provide highly enantio- and diastereoenriched
syn-cis-disubstituted cyclopropyl alcohols (Figure 5B).
24 A variety of 1-chloro-1-alkynes and aldehydes (saturated, aromatic, and heteroaromatic) were employed successfully in these tandem reactions.
Figure 5. Tandem syntheses of (A) epoxy and allylic epoxy alcohols and (B) syn-cis-disubstituted cyclopropyl alcohols.
4.2 Catalytic Asymmetric Synthesis (Z)-Trisubstituted Allylic Alcohols
The (
Z)-vinyl zinc reagents generated were
all disubstituted because the 1,2-metalate rearrangement was executed with a hydride source. A variant of the 1,2-metalate rearrangement/transmetalation sequence for the stereospecific generation of (
Z)-
trisubstituted vinyl zinc reagents was also developed.
25 Hydroboration of 1-bromo-1-hexyne with either diethyl- or dicyclohexylborane followed by 3 equiv of diethylzinc provided the (
Z)-trisubstituted vinyl zinc reagents. The dialkylzinc served a two-fold function: it induced a 1,2-metalate shift to form the new C-C bond and promoted the boron to zinc transmetallation. In the presence of TMEDA (to inhibit the zinc halide by-product) and catalytic amounts of (-)-MIB, these reagents were then employed in the catalytic asymmetric addition of (
Z)-trisubstituted vinyl zinc reagents to aldehydes to furnish enantioenriched (
Z)-trisubsituted allylic alcohols in good yields and excellent enantioselectivity (Scheme 2). Unfortunately, this procedure could not be adapted to the highly enantioselective synthesis of α-methyl-substituted allylic alcohols despite screening several additives (ee <30%, 15-50% yield).
Scheme 2. Catalytic asymmetric synthesis of α-ethyl and α-cyclohexyl (Z)-trisubstituted allylic alcohols.
Tandem Catalytic Asymmetric Addition/Diastereoselective Cyclopropanation
5.1. Synthesis of syn-Cyclopropyl Alcohols. Two tandem routes have been developed to synthesize highly enantio- and diastereoenriched
syn-cyclopropyl alcohols.
26 The first route involved enantioselective alkyl addition to α,β-unsaturated aldehydes in the presence of (-)-MIB (Figure 6A) whereas the second route involved addition of vinylzinc reagents to aldehydes to furnish the allylic alkoxide intermediates (Figure 6B). After removal of the volatile materials, the intermediate allylic alkoxide was exposed to either EtZnCH
2I or the more reactive CF
3CH
2OZnCH
2I
27 to furnish
syn-cyclopropyl alcohols with high enantio- and diastereoselectivity. The first route has a broader substrate scope, but is more challenging because unsaturated aldehydes isomerize readily. The second route provides only (
E)-disubstituted
syn-cyclopropyl alcohols.
Figure 6. Tandem asymmetric A) alkyl addition to enals followed by diastereoselective cyclopropanation and B) vinylation of aldehydes followed by diastereoselective cyclopropanation.
5.2. Synthesis of Enantioenriched Dienols and syn-Vinylcyclopropanes
The catalytic enantioselective vinylation was extended to addition of dienyl groups to aldehydes in the presence of 10 mol % (-)-MIB.
24 The requisite dienyl zinc intermediates were synthesized via chemo- and regioselective hydroboration of enynes followed by transmetallation with diethylzinc. Dienols were obtained in 79-93% yield and 76-94% ee (Figure 7A).
This methodology was further extended to the synthesis of vinylcyclopropanes (VCPs). The dienylzinc alkoxide intermediates were subjected to EtZnCH
2I to provide vinyl cyclopropanes with high chemo-, enantio- and diastereoselectivity (Figure 7B). The alkoxide directed cyclopropanation of allylic C=C bonds is faster than remote C=C bonds.
28 A limitation of this method is that aromatic aldehydes were unsuccessful coupling partners.
Figure 7. Asymmetric dienylation and diastereoselective cyclopropanation.
5.3. Synthesis of anti-Cyclopropyl Alcohols
The inherent bias for
syn-selectivity in the alkoxide directed cyclopropanation could be switched to synthesize
anti-cyclopropyl alcohols using a strategy developed by Charette and coworkers
29 wherein allylic alcohols were protected with bulky silyl groups to prevent coordination to zinc carbenoids. In our method for the synthesis of
anti-cyclopropanes,
19 we silylated the intermediate zinc alkoxide in situ with TMSCl/Et
3N and then subjected the silyl ether to cyclopropanation conditions. The cyclopropyl zinc alkoxides were desilylated in situ to furnish
anti-cyclopropyl alcohols in 60-82% yield with high enantio- and diastereoselectivity (Scheme 3). The Et
3N is likely necessary to break up the zinc aggregates, rendering the zinc alkoxides more nucleophilic towards TMSCl.
Scheme 3. One-pot tandem asymmetric synthesis of anti-cyclopropyl alcohols.
5.4. Synthesis of syn-Halocyclopropyl Alcohols
The catalytic enantio- and diastereoselective tandem generation of cyclopropyl alcohols was extended to the synthesis of halo-substituted cyclopropyl alcohols.
26,30 The enantioenriched zinc alkoxide intermediate was subjected to Et
2Zn, CF
3CH
2OH, and either iodoform, bromoform or dichlorobromomethane to furnish iodo-, bromo- or chlorocyclopropyl alcohols respectively in good yields and excellent enantioselectivity (Figure 8A). In these one-pot tandem halocyclopropanation reactions, four consecutive stereogenic centers are established with excellent diastereoselectivity starting from simple achiral α,β-unsaturated aldehyde precursors. Interrogation of the cyclopropyl stereochemistry via
1H NMR and X-ray analyses led to an interesting find; when R
4 = alkyl or H, the halo group was
cis to the carbinol, whereas when R
4 = Ph, the halo group was
trans. This switch in stereochemical bias was rationalized by invoking a zinc-phenyl-p interaction.
31A complementary approach to iodocyclopropyl alcohols was developed using MIB-catalyzed asymmetric vinyl addition as the first step followed by subjection of the zinc alkoxide intermediate to 3 equiv of Zn(CHI
2)
2 (Figure 8B).
30 The iodocyclopropyl alcohols can be further allylated with allyl/methallyl bromide in the presence of LiCu(
n-Bu)
2 to furnish 1,2,3-disubstituted cyclopropanes in good yields as single diastereomers with full retention of the cyclopropane stereochemistry (not shown).
Figure 8. Tandem asymmetric (A) alkyl addition to enals followed by diastereoselective halocyclopropanations and (B) vinyl addition to aldehydes followed by diastereoselective iodocyclopropanation.
Catalytic Asymmetric Aminovinylation of Aldehydes: Synthesis of β-Hydroxyenamines, β-Aminoalcohols, and syn-Aminocyclopropyl Alcohols
Regioselective hydroboration of ynamides followed by boron to zinc transmetallation and subsequent addition to aldehydes in the presence of 5 mol % (-)-MIB furnished β-hydroxyenamines in moderate yields and high enantioselectivities (up to 98% ee, Figure 9A).
32A tandem catalytic asymmetric aminovinylation/diastereoselective cyclopropanation reaction was developed to synthesize
syn-aminocyclopropyl alcohols with excellent diastereoselectivities (>20:1) in moderate yields (Figure 9B).
32Figure 9. A) Synthesis of β-hydroxyenamines and B) tandem synthesis of syn-aminocyclopropyl alcohols.
Catalytic Asymmetric Ethoxyvinylation of Aldehydes
Highly enantioselective addition of ethoxyvinyl zinc reagents, generated via hydroboration of ethoxyacetylene followed by in situ transmetalation to zinc and addition to aldehydes in presence of MIB afforded hydroxyenol ethers with high ee (89-95%) and yields (>93%, Scheme 4).
33 Subsequent hydrolysis generated two carbon homologated enantioenriched
β-hydroxy aldehydes. In the case of addition to chiral
β-hydroxy aldehydes, mismatched and matched catalyst-substrate combinations can be used to achieve moderate to good diastereoselectivities of either the
syn- (up to 3.8:1 with (-)-MIB) or
anti-diols (>9:1 with (+)-MIB).
Scheme 4. One-pot tandem catalytic asymmetric ethoxyvinylation of aldehydes.
Other uses of MIB in Organic Synthesis
8.1. Synthesis of γUnsaturated β-Amino Acid Derivatives
A catalytic enantioselective synthesis of
γ-unsaturated
β-amino acid derivatives was achieved in three steps from trityl protected 1-butyne-3-ol.
34 The enantioenriched allylic alcohols were transformed into the corresponding allylic amines via a [3,3]-sigmatropic trichloroacetimidate rearrangement, and led to
γ-unsaturated
β-amino acid derivatives with high ee after a one-pot deprotection-oxidation sequence (Scheme 5). Similar [3,3]-sigmatropic allyl cyanate-to-isocyanate rearrangement reactions were executed to access enantioenriched allylic amines en route to the syntheses of glycocinnasperimicin D and pachastrissamine (Figure 10).
35,36Scheme 5. Asymmetric synthesis of γunsaturated β-amino acid derivatives via a [3,3]-sigmatropic rearrangement.
Figure 10. [3,3]-Sigmatropic allyl cyanate-to-isocyanate rearrangement reactions en route to the synthesis of glycocinnasperimicin D.
8.2. Synthesis of Di(allyl) Ether Derivatives
Nelson and coworkers added Et
2Zn to conjugated enals in the presence of MIB followed by O-allylation of the enantioenriched zinc alkoxides to provide di(allyl) ethers in good yields (73-87%) and high ee (88-98%).
37,38,39 Subsequent olefin isomerization and Claisen rearrangement provided access to a variety of enantio- and diastereoenriched Claisen adducts (Scheme 6).
Scheme 6. Asymmetric synthesis of di(allyl) derivatives.
8.3. One-pot Catalytic Asymmetric Synthesis of Pyranones
Enantioenriched pyranones with >90% ee were prepared via a one-pot tandem asymmetric alkylation of 2-furfurals in the presence of catalytic (-)-MIB followed by oxidation with NBS (Scheme 7).
40Scheme 7. One-pot tandem asymmetric synthesis of enantioenriched pyranones.
8.4. Synthesis of Enantiomerically Enriched 1,2,4- Trioxanes
Enantioenriched allylic alcohols can be subjected to a hydroxyl directed regio- and diastereoselective photooxygenation reaction with O
2 in the presence of tetraphenyl porphyrin (TPP) to obtain allylic hydroperoxides in >10:1 dr, with the
threo-isomer as the major product (Scheme 8).
41 The allylic hydroperoxides were further reacted with cyclic ketones in the presence of catalytic
p-TsOH to yield enantioenriched 1,2,4-trioxanes (16-78% yield) that exhibited antimalarial activity.
Scheme 8. Synthesis of enantioenriched β-hydroperoxy alcohols and 1,2,4-trioxanes.
8.5. Polystyrene-supported MIB-derived Ligands
Pericas and coworkers have successfully synthesized and immobilized 3-exopiperazinoisoborneol (PIB), a close analog of MIB, to Merrifield resins (Scheme 9).
42 Polystyrene-supported PIB possessed high catalytic activity and improved chemical stability, and was employed as a ligand (10 mol %) in the asymmetric alkylation of aldehydes with Et
2Zn in batch methods to produce highly enantioenriched alcohols in good yields (50-92% y, 92-99% ee). This method is amenable to continuous flow methods for over 30 h with high conversion and no erosion in enantioselectivity. In this fashion, industrial scale amounts (13.0 g) of enantiopure alcohol were isolated in a single continuous flow operation leading to >30 fold better performance compared to batch conditions (TON = 251 with respect to the product).
Scheme 9. Synthesis of polystyrene-supported PIB
8.6. MIB in Syntheses of Natural Products
Over the years, MIB has been employed in the synthesis of a number of natural products. In all of these syntheses, MIB has been primarily used in asymmetric alkylation or vinylation reactions to provide diastereo- and enantioenriched alcohols.
35,36,41,43,44 In their formal synthesis of leucascandrolide A, Hong and coworkers employed (-)-MIB in the diastereoselective vinylation of a chiral aldehyde to furnish the requisite allylic alcohol with >32:1 diastereoselectivity (Figure 11).
44 The enantio- and diastereoselective addition step can be further coupled with efficient transformations en route to synthesis of natural products. As outlined earlier, a [3,3]-sigmatropic allyl cyanate-to-isocyanate rearrangement reaction was employed in the synthesis of glycocinnasperimicin D and pachastrissamine (jaspine B) (Figure 10)
35,36 while a diastereoselective Schenk ene reaction with singlet oxygen was utilized in the synthesis of artemisin-type 1,2,4-trioxanes (Scheme 8).
41 It is hopeful that synthetic chemists will adopt the tandem chemo-, regio- and diastereoselective transformations reactions presented herein in efficient synthesis of their natural products.
Figure 11. Hong and coworkers diastereoselective vinyl carbonyl addition in the formal synthesis of leucascandrolide A.
Both Nugent's (+)- and (-)-MIB have found significant synthetic utility as a ligand of choice for addition of organozinc groups to carbonyl compounds. The efficient installation of chirality, coupled with tandem chemo-, regio- and diastereoselective transformations, provides high-value added building blocks that augment the synthetic organic chemist's repertoire of enantioenriched small molecules.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved