Org. Synth. 2013, 90, 215-228
DOI: 10.15227/orgsyn.090.0215
Discussion Addendum for: Preparation of 4-Acetylamino-2, 2, 6, 6-tetramethylpiperidine-1-oxoammonium Tetrafluoroborate and the Oxidation of Geraniol to Geranial (2,6-Octadienal, 3,7-dimethyl-, (2e)-)
Submitted by James M.Bobbitt*
1, Nicholas A.Eddy
1, Jay J.Richardson
1, Stephanie A.Murray
2, and Leon J.Tilley
2.
Discussion
The useful nitroxide catalyst, 4-acetamido-TEMPO 2 and the oxoammonium salt, 4-acetamido-2,2,6,6-tetramethyl-1-oxopiperidinium tetrafluoroborate (4-acetylamino-2,2,6,6-tetramethylpiperidin-1-oxoammon-ium tetrafluoroborate, Bobbitt's salt) 3 have been prepared from 4-amino-2,2,6,6-tetramethylpiperidine 1 in molar amounts in an inexpensive and high yielding, revised procedure.Compound 1 is an industrial chemical available in quantity from TCI-America at a reasonable price, as well as from many other vendors.Using current prices, 2 and 3 can be prepared for less than $1.00 per gram.The procedure is shown in Scheme 1.
In addition to this revised preparation procedure (A), we will report in this addendum an overall view of stoichiometric oxoammonium chemistry (B), solubility properties of 2 and 3 and their relevance (C), the relative oxidation rates for hydroxyl groups in various chemical environments (D), examples of selective reactions (E), and some recent important developments from other laboratories (F).
A. The Revised Preparation of 2 and 3 (Scheme 1)3
Several changes have been made from the original procedure.
4-8 The acetylation of
1 to its acetamido derivative is now carried out in ice water and combined with base, catalysts, and peroxide to give in "one pot" the acetylated TEMPO derivative
2 in 90-93% yield.In the oxidation of
2 to
3 with HBF
4 followed by bleach (NaOCl), NaBF
4 is added to salt out the final product
3 in 90-93% yield.The NaBF
4 addition results in common ion assisted precipitation, which provides an enhanced yield and product purity.The reduced solubilities are shown in Table 1.The procedure is entirely "green" since the only solvent is water.
Precise instructions are given for the recrystallization of
2 and
3, if needed, and for the recovery of the leftover compounds after each step in the sequence.
3Scheme 1. Revised Procedure for the Preparation of 2 and 3.
B. Overview of Oxidations with the Oxoammonium Ion
All of these reactions have been discussed in detail in an
Organic Reactions chapter
9 and are shown in Scheme 2.The catalytic reaction (1) is certainly the most widely used since it is inexpensive and efficient.However, it is quite complex, has been discussed in detail
9 and will not be considered here.Variations on the stoichiometric reactions (2) are neutral reactions in DCM with silica gel (2a), reactions in the presence of pyridine bases (2b), and Golubev disproportionation oxidations (2c).The stoichiometric reactions are colorimetric and highly dependent on solubilities, primarily in DCM.These solubilities have been measured and are also listed in Table 1.
3Scheme 2. Overall Stoichiometric Oxidations
The stoichiometric reactions can be divided into three methods.
Method 2a.The simplest of the three methods is the neutral oxidation in DCM and silica gel.
5 The reaction is colorimetric in that a bright yellow slurry of
3 and silica gel is converted to a white slurry of the reduced oxidant
4, the product and silica gel. The yields are near quantitative.Since both
3 and
4 are only slightly soluble in DCM (Table 1), the course of the reaction can be followed by filtering samples of the reaction mixture through a cotton wad and analyzing the samples by GC, TLC or even NMR.
10 A simple filtration through a 2-4 mm pad of silica gel yields a DCM solution of pure aldehyde or ketone that can be isolated or used in a tandem reaction.The method is ideal for the preparation of small amounts of volatile or hygroscopic aldehydes or ketones for tandem reactions.The major disadvantage of the method is that aliphatic alcohols react slowly, frequently requiring one or two days.Relative rates are presented in Table 2.
Method 2b. Oxidations in the presence of pyridine bases have been less explored, but this variation may become more important.
11-13 In general, oxidations in the presence of pyridine tend to give esters,
11 and oxidation in the presence of 2,6-lutidine tend to give aldehydes from primary alcohols.Again, the reactions are colorimetric, going from a yellow slurry to a bright orange red color due to the nitroxide
2, which is the product of the reduction of
3.The reaction is fast and is a more powerful oxidation system than method 2a.For example, trifluoromethylcarbinols are oxidized to ketones using method 2b,
12 but this reaction does not take place under neutral (2a) conditions.The disadvantage of this method is that both the pyridine base and the nitroxide must be removed for product isolation.This can be accomplished in several ways.
11-13
Method 2c. Method 2c depends upon the
p-toluenesulfonic acid-mediated disproportionation of two equivalents of
2 into a mixture of tosylate salts of
3 and
4, which is the Golubev reaction.
14 The oxidation is carried out by the oxoammonium salt to give product and tosylate
5.This method has been extensively used.
4,9 Once again, the reaction is colorimetric in that the yellow slurry is converted to the white reduced form, a tosylate salt
5. This salt precipitates, but the precipitate is somewhat more soluble than the tetrafluoroborate salt (Table 1).The reaction is fast, but sometimes the products require further purification.
4
C. Solubility Studies and Colors of Oxoammonium Salts and Related Products
The solubility properties and the colors of the various compounds are important for the understanding of the reactions and are given in Table 1.The general discussion is given in the preceding section.
Table 1. Solubilities and Colors of the 4-Acetamido Compounds.
|
|
Water |
DCM (rt) |
Diethyl Ether |
Color |
|
| 2 |
3 g/100 mL at 0 ºC |
soluble |
0.28 g/100 mL at rt |
orange |
| 3.5 g/100 mL at rt |
|
|
|
| >50 g/100 mL at 100 ºC |
|
|
|
| 1 g/100 mL in 2 M NaCla |
|
|
|
|
| 3 |
6 g/100 mL at 0 ºC |
0.1 g/100 mL |
insoluble |
yellow |
| 8 g/100 mL at rt |
|
|
|
| >50 g/100 mL at 100 ºC |
|
|
|
| 1.8 g/100 mL in 0.8M NaBF4a |
|
|
|
|
| 4 |
soluble |
0.02 g/100 mL |
insoluble |
white |
|
aThese data are important for the revised preparation of 2 and 3.3
D.Rate Studies of Type 2a Reactions The rates of oxidations by method 2a are given in Table 2.The reference compound is benzyl alcohol.The rates were measured by oxidizing a mixture of two alcohols with half of the theoretical amount of oxidant followed by NMR analysis.These data are accurate to only one significant figure and are taken from two papers.
5,15
Table 2. Relative Rates of Oxidation of Various Alcohols with 3 Compared to the Oxidation of Benzyl Alcohol (rate = 1.0).
E.Examples of Selective Alcohol Oxidations of Polyalcohols
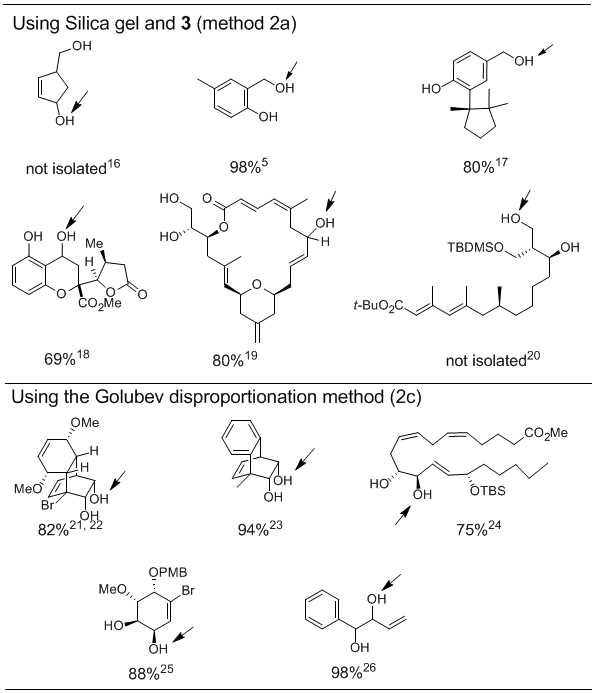
As shown above, changes in the substitution pattern of alcohols result in different rates of oxidation, and this selectivity can be quite useful (see Table 3).In general, allyl and benzyl alcohols can be oxidized selectively in the presence of aliphatic alcohol groups, and less sterically hindered aliphatic alcohols are easier to oxidize than more hindered alcohols.For each example the site of selective oxidation is indicated by an arrow, and the yield of the oxidation and primary reference are given below the structures.
Table 3.Selective Oxidations of Polyalcohols
F.Recent Literature and Some Unique New Reactions Several recent reviews of oxoammonium salt and nitroxide-catalyzed oxidations have been published.
6,9,27,28 The following reactions are examples of some new chemistry of oxoammonium salts (other than the traditional oxidations of alcohols to aldehydes or ketones) which have been published since our
Organic Reactions review (Miscellaneous Reactions on page 160).
9 Only one specific reaction is given in each case, although a number of similar substrates are described in each reference. In entry 1, oxidations in water are described.
29 In entries 2 and 3, methods for direct oxidation of alcohols to carboxylic acids are given.
15,30 Entry 2 is of special interest, because an oxoammonium salt is used as a catalyst for oxidation rather than the usual nitroxide. In the reactions shown in entry 4,
15 the remarkable selectivity of oxoammonium salts is illustrated: a benzyl alcohol can be oxidized in 98% yield in the presence of an aliphatic primary alcohol; in the second reaction, a primary alcohol can be oxidized to a dialdehyde in 97% yield; and in the last equation, an aliphatic aldehyde can be oxidized to a carboxylic acid in 95% yield in the
presence of an aromatic aldehyde.
The synthesis of α,β-unsaturated ketones from silylenol ethers is described in entry 5.
31 Entry 6 involves a use of an oxoammonium salt as a cleavage reagent for benzyl ethers.
32 In entry.7, a unique addition of an adjacent keto group to a cyclic ketone followed by the formation of a double bond is shown.
33 Entries 8, 9, and 10 show the imaginative use of metal ions as catalysts for interesting one-pot oxidations to form new carbon-carbon bonds.
34-36 In entry 11, a "one-pot" reaction similar to entries 8-10 is shown with only an oxoammonium salt and no metals.
37 In entries 12 and 13, the lutidine conditions (Scheme 2, eq 2b) for oxoammonium salt oxidation are presented.Neither reaction takes place under neutral conditions (Scheme 2, eq 2a).Vinyl, aryl, propargyl and aliphatic trifluoromethylcarbinols (entry 12) were oxidized to the trifluoromethyl ketones in fair to excellent yield.
12 Entry 13 shows the oxidation of a complex neopentyl alcohol, which was oxidized under these conditions.
38
Table 4. New Chemistry of Oxoammonium Salts
Table 4 (cont)
 |
James M.Bobbitt was born in Charleston, WV in 1930.He received the B.Sc.in chemistry from West Virginia University in 1951 and the Ph .D.degree from The Ohio State University in 1955 with M.L.Wolfrom.He joined the University of Connecticut in 1956 after a year of postdoctoral work with Carl Djerassi.He started his career working on the structure elucidation and synthesis of natural products, then shifted to electroorganic oxidations with the intent to mimic natural synthetic reactions.In 1985, he discovered oxoammonium salt oxidations.In 1992, he formally retired from UCONN and since then has carried out bench chemistry in oxidation reactions.
|
 |
Nicholas Eddy was born in Johnstown, PA in 1980.He obtained his B.S.degree in Chemistry from Indiana University of Pennsylvania in 2003, and M.S.degree in Chemistry in 2006 at Indiana University of Pennsylvania.He went on to pursue his Ph.D.in Chemistry at the University of Connecticut under the direction of Dr.Gabriel Fenteany, which was completed in November 2012.During his graduate studies, he worked on developing an oxidation with oxoammonium salts of cyclic 1,3-diketones leading to cyclohex-5-ene-1,2,4-triones.
|
 |
Jay Richardson was born in Providence, RI in 1990.He studied chemical engineering at the University of Connecticut, where he pursued undergraduate research under the direction of Dr.Gabriel Fenteany and Nicholas Eddy.After completing his B.S.degree, he began work for Zeeco Corp.as a combustion research engineer.
|
 |
Stephanie A.Murray was born in Holyoke, MA in 1991.She is currently working toward a B.S.degree in Chemistry at Stonehill College in Easton, MA which she expects to receive in May 2013.Under the direction of Dr.Leon Tilley, her undergraduate research focuses on trifluoromethyl directed gamma-silyl elimination in the cationic mediated synthesis of trifluoromethylcyclopropanes.She also spent time in the Hartwig group at the University of California, Berkeley where she worked on palladium-Josiphos catalyzed benzamide synthesis and rhodium-NHC catalyzed hydroamination of olefins.
|
 |
Leon J.Tilley was born in New Haven, CT in 1968.He received a B.A.in chemistry and a B.A.in Russian language from Grinnell College, in Grinnell, Iowa in 1990 and a Ph.D.in chemistry from Indiana University, Bloomington in 1996.He joined the faculty at Stonehill College in 1996.His research interests include: silyl substituted cations; synthesis of strained hydrocarbons; fluoroalkyl compounds; oxidation reactions; and highly symmetric molecules.In 2011, he prepared 1-(trifluoromethyl)bicyclo[1.1.0]butane.He and his undergraduate students, in collaboration with colleagues and graduate students at UCONN are currently preparing a variety of trifluoromethyl- substituted strained hydrocarbons.Professor Tilley wishes to acknowledge the Office of Naval Research (ONR) (Award No.: N00014-11-1-0921) for partial funding of this work.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved