The discovery of
BINAP by Noyori and co-workers in 1980 ushered in an era of fertile research and practical applications of asymmetric catalysis using axially-chiral phosphine ligands.
2 The first practical syntheses of
(R)- and
(S)-BINAP (
4) were reported by Takaya
et al in 1986 (Scheme 1), involving synthesis of the racemic bis-phosphine oxide, resolution with
(R,R)-dibenzoyl tartaric acid, followed by reduction to the bis-phosphine.
3 While this route supported production of kilogram quantities of
BINAP, some drawbacks were the harsh conditions (320 °C) and low yield (45%) for conversion of
1,1'-bi-2-naphthol (
BINOL) to the dibromide and a resolution in which the (
S)-isomer could be isolated in high ee with
(R,R)-dibenzoyl tartaric acid while the opposite enantiomer required an upgrade with
(S,S)-dibenzoyl tartaric acid after recovery from the supernatant.
BINOL Resolution
A more concise approach to
BINAP would be to start with resolved
BINOL and develop mild conditions for conversion to
BINAP such that the chiral integrity is not compromised. A number of viable methods for resolving
BINOL have been developed over the past 40 years, including enzymatic hydrolysis of its diester, resolution of its
phosphoric acid, and formation of a variety of diastereomeric salts and co-crystals with chiral bases.
4 Given the wide availability of the
Cinchona alkaloids, we decided to further investigate the resolution of
BINOL discovered by Toda
5using
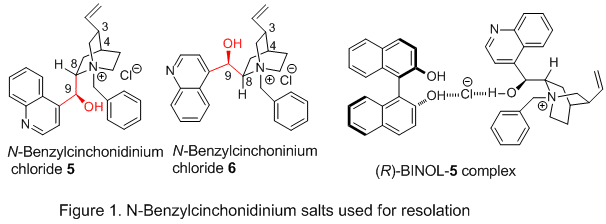
N-benzylcinchonidinium chloride 5 (Figure 1). This alkaloid forms a salt co-crystal with
(R)-BINOL with the chloride serving as a bridge between the alkaloid and
BINOL with three hydrogen bonds: one to an OH of
BINOL, one to the OH of
5, and a third to an OH of a second molecule of
BINOL.
6For most applications that employ
Cinchona alkaloids, the two "pseudo-enantiomeric" pairs,
cinchonine/
cinchonidine and
quinine/
quinidine, lead to opposite product enantiomers since the pairs have opposite stereochemistry at the "working" part of the molecule, C8 and C9. For example, in the first practical resolution of
BINOL, the phosphoric acid of
BINOL is resolved using
cinchonine and
cinchonidine to access the (+)- and (-)-enantiomers, respectively.
7 Surprisingly, however,
N-benzylcinchonidinium chloride 5 and
N-benzylcinchoninium chloride 6 both form salt co-crystals only with
(R)-BINOL.
8 Thus, to efficiently isolate both pure enantiomers of
BINOL using either
5 or
6, a near perfect resolution is required, with one diastereomeric complex crystallizing in very high yield, leaving the other enantiomer with high enantiomeric purity in the supernatant.
In the published procedure of Toda,
6 MeOH was used as the crystallization solvent, affording
(R)-BINOL in high yield and ee (after breaking the complex) but leaving the (
S)-enantiomer in the supernatant in only 42% ee. Thus, the pure (S)-enantiomer could not be isolated via this method. We initiated our studies of this resolution by undertaking a survey of solvents and identified
MeCN as the most promising:
5 had reasonable solubility, the crystalline diastereomeric complex
(R)-BINOL•
5 had very low solubility at 0 °C, and no evidence was found for crystallization of
BINOL•
5 in this solvent. A straightforward resolution for isolating both enantiomers of
BINOL in high yield was thus developed.
9The resolution was carried out with 0.55 equiv of 5 at a concentration of 80 g of BINOL per L of solvent. The mixture was refluxed for 4 h to ensure BINOL and 5 were fully dissolved, during which time crystallization of the (R)-BINOL•5 complex occurred. After cooling to 0 °C, the solids were filtered and washed with MeCN to afford (R)-BINOL•5 co-crystals with 96% de. The complex was upgraded to >99% de by slurrying in MeOH, then (R)-BINOL was recovered by salt break in EtOAc/aq. HCl. Concentration of the EtOAc layer to dryness afforded (R)-BINOL (85-88% recovery, >99% ee).
The supernatant from the original crystallization, which contained BINOL, was concentrated to dryness, then dissolved in EtOAc and washed with dilute HCl to remove any remaining 5. Concentration of the EtOAc layer to dryness afforded BINOL (89-93%, recovery, 99% ee). This straightforward resolution thus provides access to each enantiomer of BINOL in =99% ee with recoveries in the 85-93% range for each enantiomer.
Nearly concurrent with our 1995 publication, Pu reported a resolution with
5, isolating each enantiomer of
BINOL in 70-75% yield.
10 This group carried out the initial resolution in
MeOH to crystallize
(R)-BINOL•
5. The supernatant was evaporated and the resulting solids slurried in
EtOAc, affording additional crystalline
(R)-BINOL•
5 adduct, with the supernatant containing
BINOL with 84% ee. The material from the supernatant was concentrated then further upgraded via crystallization from
benzene.
Three analogs of
BINOL have been similarly resolved, 6,6'-dimethyl and 7,7'-dimethoxy using
511 and 7,7'-bis(benzyloxy) using
quinine.
12A 2005 review indicates the resolution with
N-benzylcinchonidinium chloride described in our 1999
Organic Syntheses procedure remains among the most effective and straightforward methods of accessing both enantiomers of
BINOL.
4BINAP Synthesis
In parallel to the development of the
BINOL resolution, a more efficient route to
BINAP was explored. Five papers in the early 1990's provided the starting point for our efforts. Activation of
BINOL as its bis-triflate
3 was reported by Mattay in 1990,
13 then Morgans
14a and Hayashi
14b discovered that mono-phosphorylation of bis-triflate
3 could be accomplished using
Ph2P(O)H and
Pd catalysis, noting the reaction occurred stereospecifically with no loss of ee. In addition, cyanation could also be achieved using
nickel catalysis, affording primarily mono-substituted product with up to 10% of the bis-nitrile.
14a Snieckus reported conversion of bis-triflate
3 to the bis-methyl derivative using
MeMgBr with
Ni catalysis.
15 While no aryl triflate to aryl phosphine couplings were known at the time we began our work, cross-couplings of aryl iodides with phosphine-boranes, prepared from the corresponding phosphine oxide and LiAlH
4-NaBH
4, were reported by Imamoto using
Pd catalysis.
16In considering the direct cross-coupling of diphenylphosphine with bis-triflate
3, a transition-metal catalyst must be chosen that will not be poisoned by ligation to the phosphine ligands present in the starting material and product. The 2
nd and 3
rd row transition metals such as
Pd,
Rh,
Ru, and
Pt were thus ruled out, with
Ni identified as having the best probability of success given its "hardness" and likely only weak ligation to phosphines. Screening identified a number of
Ni catalysts that gave some conversion to product, with
NiCl2(dppe) selected for further development. The optimized procedure used 10 mol % catalyst, 4 equiv
DABCO, 2.4 equiv
Ph2PH and
DMF as solvent. The reaction required 2-3 days at 100 °C to reach completion.
BINAP crystallized directly from the reaction mixture, providing product in 77% yield (97 % purity (HPLC area), >99% ee).
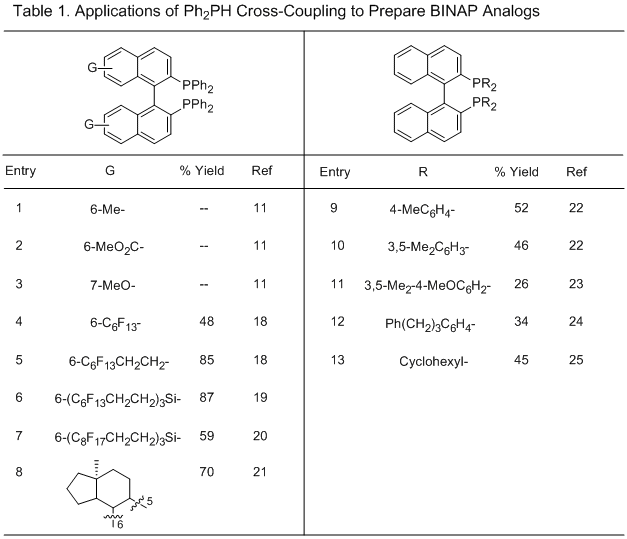
17Since our original report in 1994, applications of this methodology to several
BINAP analogs for various applications have been reported (Table 1). The 6-Me, 6-MeO
2C, and 7-MeO analogs (Table 1, entries 1-3) were prepared according to the same procedure except the cross-coupling reactions were completed overnight rather than 2-3 days.
11 Several fluorous analogs have been prepared in 48-87% yields (Table 1, entries 4-7).
18, 19, 20 The bis-steroid ligand (Table 1, entry 8) was readily prepared from the natural product equilenine. The resulting diastereomers could be separated by column chromatography, eliminating the need for a resolution.
21 The electron-rich BINAP analog (Table 1, entry 11) was prepared in a modest 26% yield, but was found to afford superior ee's in the Rh-catalyzed asymmetric 1,4-addition of arylboron reagents to 5,6-dihydro-2(
1H)-pyridones to prepare 4-aryl-2-piperidones.
23 While most of the coupling partners in Table 1 are arylphosphines, Wills reported preparation of the bis(dicyclohexylphosphine) analog in 45% yield from
Cy2PH by a modified procedure utilizing Zn dust as an additional reagent (Table 1, entry 13).
25 Attempts to prepare the bis(di-
t-butylphosphine) analog were unsuccessful, with only the mono-substitution product formed.
25 Gilheany also reported formation of the mono-substituted product using
t-BuPhPH in 40% yield.
26 Ni- and
Pd-catalyzed cross-couplings of Ar
2PH with triflates to prepare mono-phosphine BINAP analogs, as well as non-BINAP structures, have also been reported but are outside the scope of this addendum.
27
Alternate Methods to Prepare BINAP and Analogs27
A number of alternate methods of preparing BINAP and analogs have been reported in the past two decades.
Cross Coupling of bis-Triflate 3 with Other Phosphorus Partners (Scheme 2). Takasago scientists reported a synthesis of
BINAP via cross-coupling of bis-triflate
3 with Ph
2P(O)H, citing the ease of handling the
phosphine oxide vs the phosphine (pyrophoric, stench) as the principle driver.
28 Employing the same
NiCl2(dppe) catalyst that was used in the Merck procedure, the cross-coupling affords a mixture of
BINAP analog
9 (35%), the mono-oxide
8 (60%), and the bis-oxide (4%). The mixture is then treated with HSiCl
3/PhNMe
2 to reduce all species to the
BINAP analog. This method has been applied to a number of analogs, including 2-napthyl, 4-tolyl, 3,5-dimethylphenyl, 4-methoxy-3,5-dimethylphenyl, and 4-
t-butoxy-3,5-dimethylphenyl. Yields from
BINOL range from 57-84%.
28
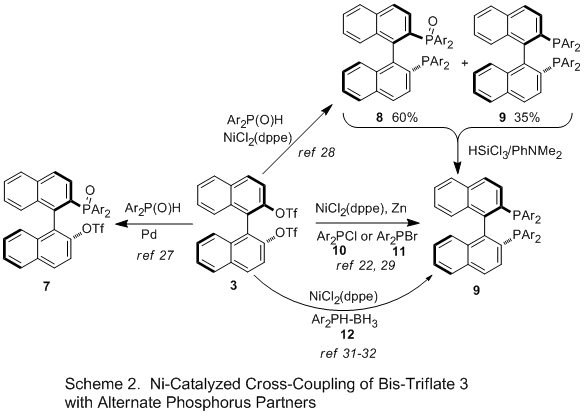
Cross-coupling of bis-triflate
3 with Ar
2P(O)H and R
2P(O)H using
Pd-catalysis stop at the mono-phosphine oxide
7, leaving an unreacted triflate that can be further functionalized using
Pd- or
Ni-catalysis to prepare unsymmetrical
BINAP analogs.
27Also using
NiCl2(dppe) catalyst, Laneman at Monsanto reported coupling of the bis-triflate
3 with
Ph2PCl, mediated by 1.2-1.5 equiv Zn, affording a yield of
BINAP of 52%.
29 The authors proposed that Zn plays two roles, reducing Ni(II) to Ni(0) as well as generating Ph
2PZnCl as the active species. This process has been reportedly licensed to Rhodia for commercial production.
27b In a slight variation of this process, Waechtler used the -SO
2C
4F
9 analog for the cross-coupling with
Ph2PCl.
30Yu found that
BINAP analogs with electron-rich aromatic groups could be prepared by the Merck process with bis-triflate
3 in moderate yields (Table 1, entries 9, 10), but that electron-withdrawing analogs were unreactive. However, using Ar
2PBr
11 instead of the chloride
10, the method could be extended to 4-CF
3 (33% yield) and 3,5-(CF
3)
2 (28% yield) analogs and yields of the electron-donating analogs could be increased 10-15%.
22A wide-variety of electron-rich phosphines have been prepared by cross-coupling phosphine borane species
12 with bis-triflate
3 in yields ranging from 24-79%.
31, 32 The use of conditions and reaction times very similar to the Merck process suggest the reaction may be proceeding via a common intermediate.
Cross Coupling of bis-Bromide with Ar2PBr. Yu reported the racemic bis-bromide of
BINOL could be converted to the corresponding Grignard species and cross-coupled with Ar
2PBr, requiring no catalyst. Five examples were reported, affording yields of 68-78% for both electron-withdrawing and donating groups. Under these conditions, Ar
2PCl was unreactive.
22
Oxidative Homo-Coupling. Takasago scientists designed and prepared a family of ligands termed SEGPHOS, which have a smaller dihedral angle than
BINAP and show superior performance in a number of applications. The preparation of this ligand is shown in Scheme 3, featuring an iron-mediated homo-coupling to generate the bis-phosphine oxide
(R,S)-15, followed by resolution to
(R)-15, and reduction to
(R)-SEGPHOS (
16).
33
In summary, axially-chiral ligands, introduced by Noyori and co-workers in 1980, have played an important role in the development of asymmetric catalysis for organic synthesis. A number of practical routes to BINAP and analogs have made a wide variety of these ligands available for application in academic settings as well as industrial processes.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved