Org. Synth. 2014, 91, 72-82
DOI: 10.15227/orgsyn.091.0072
Palladium-catalyzed 1,4-Addition of Terminal Alkynes to Conjugated Enones
Submitted by Feng Zhou, Liang Chen and Chao-Jun Li
1*
Checked by Lan-Ting Xu and Dawei Ma
1. Procedure
7-Phenyl-6-heptyn-3-one. An oven-dried, 100-mL, three-necked, round-bottomed flask is equipped with a nitrogen inlet adapter, a stopcock, a rubber septum and a Teflon-coated magnetic stir bar (22 mm x 8 mm) at room temperature under a flow of nitrogen. While temporarily removing the septum, the flask is charged with Pd(II) acetate (Pd(OAc)2) (0.224 g, 1 mmol, 0.02 equiv) (Note 1) and trimethylphosphine (PMe3) (4 mL of 1 M PMe3 solution in toluene, 4 mmol, 0.08 equiv) (Note 2) under nitrogen. The mixture is stirred at room temperature for 10 min, resulting in a homogeneous brown solution (Note 3). Deionized water (50 mL) is added (Note 4); then phenylacetylene (3.07 g, 30 mmol, 0.6 equiv) (Note 5) and ethyl vinyl ketone (6.31 g, 75 mmol, 1.5 equiv) (Note 6) are added successively via syringe (Note 7). The solution is stirred vigorously (Note 8). With the flask equipped with a stopper, additional phenylacetylene (2.04 g, 20 mmol, 0.4 equiv) is added slowly at a rate of 0.4 mL/min. At the same time, the flask is heated in an oil bath and kept at 60 °C (oil bath temperature) for 48 h with the color of the solution gradually becoming darker during the course of the reaction (Note 9). Upon completion of the reaction the color of the solution turns dark brown. The reaction mixture is allowed to cool to room temperature, transferred to a 250-mL separatory funnel, and extracted with ethyl ether (3 × 60 mL) (Note 10). The organic layer is separated and washed with saturated brine (3 × 5 mL) (Note 11). The combined organic phases are then dried over 25 g of Na2SO4 (Note 12) for 8 h. After the solid Na2SO4 is separated via gravity filtration on a funnel padded with cotton and washed with (3 × 30 mL) of ethyl ether, the filtrate is concentrated at room temperature (25 °C) by rotary evaporation (200 mmHg and then at 10 mmHg) to provide the residue (Note 13). This residue, a dark-brown oil, is adsorbed onto silica (6 g) to form a dry sample, loaded on a silica gel column, and purified by flash column chromatography (Note 14) to afford 6.85–6.94 g (75%) of the product as a brown liquid. This liquid appears to be pure by 1H and 13CNMR spectra (Note 15). Further purification is achieved by distillation at reduced pressure (120 °C/ 1 mmHg) (Note 16), which provided the product as a colorless liquid (6.20 g, 66%) (Note 17).
2. Notes
1.
Pd(OAc)2 (98%) was purchased from the Aldrich Chemical Company and used as received.
2.
PMe3 (1.0
M solution in toluene) was purchased from the Aldrich Chemical Company and used as received. This reagent should only be used under an inert atmosphere.
3.
Pd(OAc)2 may not dissolve completely.
4. The use of nitrogen gas is no longer necessary after the addition of deionized water.
5.
Phenylacetylene (98%) was purchased from the Aldrich Chemical Company and used as received.
6.
Ethyl vinyl ketone (97%, stabilized with BHT) was purchased from the Aldrich Chemical Company and used as received. It is a highly volatile reagent (bp 103-105 °C/760 mmHg) and 1.5 equiv of
ethyl vinyl ketone was used.
7. The solution became a pale-yellow emulsion after the addition of
phenylacetylene and
ethyl vinyl ketone.
8. The vigorous stirring (1400 rpm) is required to reduce the self-coupling of the
phenylacetylene.
9. The progress of the reaction was monitored by TLC analysis on silica gel with 10%
EtOAc-hexanes as eluent and visualization under the UV light as well as alkaline
KMnO4 solution. R
f (self-coupling of
phenylacetylene): 0.61; R
f (product): 0.23. The TLC plates were purchased from the EMD Chemicals Inc. (an affiliate of Merck) and were used as received.
10.
Ethyl ether (99.9%) was purchased from the Fisher Scientific.
11.
Sodium chloride (99.0%) was purchased from ACP Chemicals.
12.
Sodium sulfate (certified anhydrous) (99.4%) was purchased from the Fisher Scientific.
13. An
ethanol-cooled recirculator was used to condense the solvent and
ethyl vinyl ketone in the collecting flask. The submitters used a dry-ice acetone to provide this cooling.
14. The silica gel was purchased from the Silicycle Inc. with the particle size as 40-63 mm (230-400 mesh). Flash column chromatography was performed using silica gel (3 cm diameter × 30 cm height), eluting with 500 mL of hexanes/ethyl acetate (50/1) first to afford the byproduct
1,4-diphenyl-1,3-butadiyne (0.225 g, 4%) (
1H NMR (400 MHz, CDCl
3) δ: 7.56-7.53 (m, 4 H), 7.38-7.32 (m, 6 H);
13C NMR (100 MHz, CDCl
3) δ: 132.5, 129.2, 128.4, 121.8, 81.5, 73.9 and then hexanes/ethyl acetate (15/1) to elute the product. The collected fractions (totaling 500 mL) were analyzed using TLC (hexanes/ethyl acetate = 10/1)
(Caution! A small amount of the impurity may co-elute with the product)
. The spots were visualized using UV light and an alkaline
KMnO4 solution.
15. The physical properties are as follows:
1H NMR
pdf(500 MHz, CDCl
3) δ: 0.99 (t,
J = 7.0 Hz, 3 H), 2.38 (q,
J = 7.5 Hz, 2 H), 2.55–2.59 (m, 2 H), 2.62–2.65 (m, 2 H), 7.16–7.19 (m, 3 H), 7.26–7.29 (m, 2 H);
13C NMR
pdf(125 MHz, CDCl
3) δ: 7.7, 14.1, 36.0, 41.2, 80.9, 88.7, 123.7, 127.7, 128.2, 131.6, 209.3; IR (neat) cm
-1: 3055, 2976, 2239, 1715, 1481, 1363; GC-MS (Relative Intensity)
m/z: 186 ([M
+], 31), 171 (12), 157 (100), 128 (65), 115 (70), 102 (13), 89 (10).
16. The product obtained by column chromatography was transferred into a 25-mL round-bottomed flask equipped with a magnetic stirbar. The product was distilled under vacuum through a water-cooled condenser topped with a short path distillation head to afford a colorless liquid (oil bath temperature gradually increased from 25 to 140 °C). This compound is stable toward air and moisture and can be stored at room temperature.
17. Purity analysis data on distilled material are as follows: Anal. Calcd. for C
13H
14O: C, 83.83; H, 7.56. Found: C, 83.33; H, 7.58; HPLC > 99% area % purity at 254 nm detection (HPLC conditions, Agilent
TM 20RBAX RX-SiL column (4.6 × 250 mm), 5 µM particle size; 0.70 mL/min flow; eluent (hexanes/isopropanol = 98/2); product elutes at 5.42 min. (HPLC conditions in submitter’s report: Supelcosil
TM LC-PAH C18 column (4.6 × 250 mm), 5 µM particle size; 0.75 mL/min flow; eluent (hexanes/isopropanol = 99/1); product elutes at 5.2 min.) The sample was prepared by dissolving 5 µL of the product in 5 mL of hexanes and the injection volumes equal to 50 µL.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The development of palladium-catalyzed C–C bond formation reactions has dramatically advanced the ‘‘state-of-the-art’’ of organic synthesis.
2 The well known palladium catalyzed C–C bond formation reactions include the Heck reaction,
3 the Stille reaction,
4 the Suzuki reaction,
5 the Trost–Tsuji reaction,
6 and the Sonogashira-coupling,
7 to name a few. On the other hand, an addition reaction is an atom-economical way to construct more complex molecules from simpler units.
8Recently, increased interest has been shown in the addition of terminal alkynes to those compounds that involve sp
2 carbon, such as C=O bonds
9 or C=N bonds.
10 However, only a few examples of the addition of terminal alkynes to C=C bonds have been reported.
11 Although palladium is one of the most commonly used metals for the purpose of catalysis, palladium-catalyzed conjugate addition of alkynes to enones has not been reported prior to our work.
12We hypothesized that the absence of this method could be attributed to either (1) the facile homo- or heterodimerization of terminal alkynes (a well-known, synthetically useful process)
13 to form by-products or (2) a lower reactivity of the alkynyl palladium intermediate towards enones. Conceivably, such obstacles can be overcome by tuning the electronic properties of the ligands to coordinate with palladium.
As part of a continued interest in developing organic synthesis in water,
14 herein a simple and highly efficient Pd-catalyzed addition of a terminal alkyne to a C=C double bond of a conjugated enone, either in water or in acetone under an atmosphere of air, was achieved.
15Using the procedure described herein, various of 1,4-addition products can be easily synthesized from the terminal alkynes and conjugated enones in the presence of catalytic amounts of Pd(OAc)2 and PMe3. Table 1 lists several examples of these products. Alkynes bearing silyl, alkenyl, aromatic, aliphatic or halide all reacted smoothly with vinyl ketone to afford good yields of the desired products. With diyne as a substrate, a bis-addition adduct was achieved as a major product. In addition to ethyl vinyl ketone, methyl vinyl ketone also participated in this addition reaction, albeit in a lower yield. It should be noted that both water and acetone are effective as solvents and similar results were obtained in either solvent.
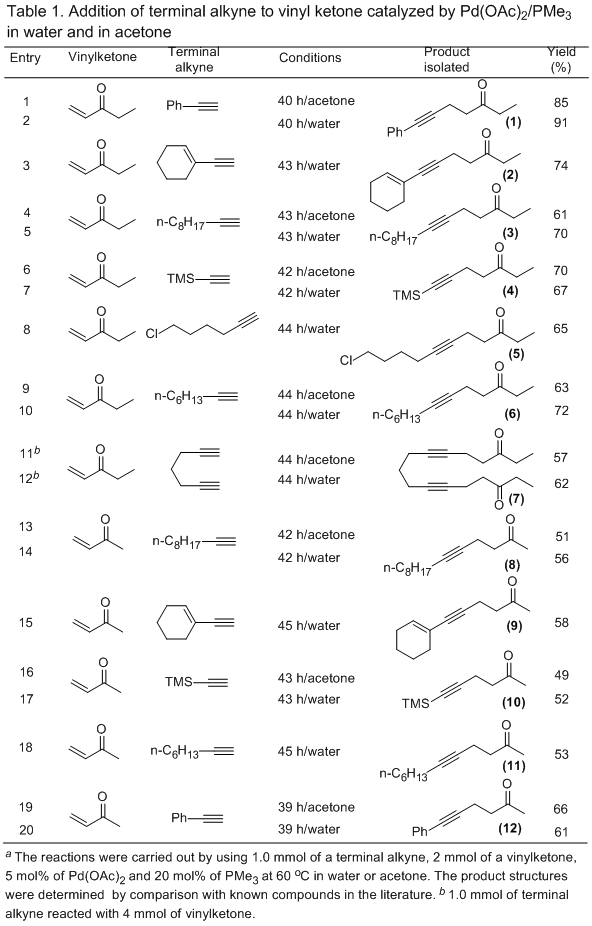
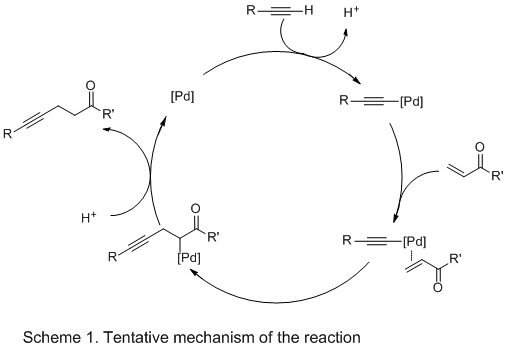
In the tentative mechanism in Scheme 1, the η
2-coordination of the triple bond to the palladium center followed by a direct deprotonation of the coordinated terminal alkyne by the palladium catalyst
16 generates the alkynyl-palladium intermediate. Then, η
2-coordination of C=C double bond to the palladium center followed by the carbopalladation,
17 and the substitution of Pd with hydrogen (either from the solvent or terminal alkyne) produces the γ, δ-ynone product with concomitant regeneration of the Pd catalyst.
In summary, the first palladium-catalyzed 1,4-addition of terminal alkynes to the C=C double bond of conjugated enones was developed in water and in acetone, under an atmosphere of air. The corresponding γ, δ-alkynyl ketones were obtained in high yields. The process is simple and can generate a wide range of alkynyl ketones.
Appendix
Chemical Abstracts Nomenclature
(Registry Number)
Phenylacetylene: Benzene, ethynyl-: (536-74-3)
Ethyl vinyl ketone: 1-penten-3-one: (1629-58-9)
Palladium (II) acetate: (3375-31-3)
Trimethylphosphine: Phosphine, trimethyl- (594-09-2)
7-Phenyl-6-heptyn-3-one: 6-Heptyn-3-one, 7-phenyl-: (185309-04-0)

|
Dr. Feng Zhou was born in 1983 in Wuhan, China. He received his B.S. degree from Wuhan University in 2006 and Ph.D. at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences in 2011 under the supervision of Prof. Xiyan Lu. He is currently working as a postdoctoral fellow with Prof. Chao-Jun Li at the McGill University in Canada, focusing on the Cross-Dehydrogenative-Coupling (CDC) react-ions via C-H activations.
|

|
Dr. Liang Chen received his B.S. degree in Environmental Science (minor: Economics) in 1999 at Nanjing University. He studied chemistry at Tulane University, where he received his Ph.D. degree in 2005 under the supervision of Professor Chao-Jun Li. He then became a Postdoctoral Researcher at Stanford University, and subsequently a Research Associate at LSU Dental School. Since 2008, he has been working in Bisco (Dental) Inc. as a Research Scientist and Senior Research Scientist.
|

|
Chao-Jun Li received his Ph.D. at McGill University (1992). After a two year NSERC Postdoctoral position at Stanford University, he became Assistant (1994), Associate (1998) and Full Professor (2000) at Tulane University. In 2003, he became a Canada Research Chair (Tier I) in Organic/Green Chemistry and a Professor (E. B. Eddy Chair since 2010) of Chemistry at McGill University in Canada. His current research efforts are focused on developing innovative and fundamentally new organic reactions that will defy conventional reactivities and have high synthetic efficiency.
|

|
Dr. Lan-Ting Xu received her BS degree from West China School of Pharmacy, Sichuan University in 2008, and her Ph.D. degree from Fudan University in 2013, under the supervision of Dawei Ma. She is now a MSD China R&D Postdoc Research Fellow in Shanghai Institute of Organic Chemistry. Her research interests include copper-catalyzed coupling reactions, metal-catalyzed direct C-H functionalization and heterocycle synthesis.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved