Org. Synth. 2021, 98, 28-50
DOI: 10.15227/orgsyn.098.0028
Chiral Organoiodine-catalyzed Enantioselective Oxidative Dearomatization of Phenols
Submitted by Muhammet Uyanik, Shinichi Ishizaki, and Kazuaki Ishihara*
1Checked by Tao Wang and Kevin Campos
1. Procedure (Note 1)
A. 3-(4-Chloro-1-hydroxynaphthalen-2-yl)propanoic acid (2). An oven-dried, 200 mL, two-necked (main 24/40, side 15/25 joints), round-bottomed flask is charged with 4-chloro-1-naphthol (7.14 g, 40.0 mmol, 1.0 equiv) (Note 2) under an air atmosphere, and the flask is equipped with a 3.5 cm, Teflon-coated magnetic stir bar, a reflux condenser fitted with a nitrogen inlet adapter, and a rubber septum (Note 3) (Figure 1A). Via the nitrogen inlet, the round-bottomed flask is evacuated under high vacuum (5.0 mmHg, few seconds) and refilled with nitrogen (three cycles). Toluene (40 mL) (Note 4), trifluoromethanesulfonic acid (0.35 mL, 4.0 mmol, 10 mol%) (Note 5) and acrylic acid (13.7 mL, 200 mmol, 5.0 equiv) (Note 6) are added via a syringe in less than 1 min each through the rubber septum. The reaction mixture is further degassed by bubbling N2 via a metal needle (16 gauge) for 20 min at room temperature with stirring. The flask is immersed in a silicone oil bath and the bath is heated to 130 °C (Figure 1B). The mixture is refluxed with stirring for 24 h under a nitrogen atmosphere (Note 7). The resulting purple solution (Figure 1C) is cooled to 0 ºC in an ice-bath for 10 min (Figure 1D). The reflux condenser is removed, and a 250 mL addition funnel is attached. An aqueous solution of K2HPO4 (120 mL, 25 wt%) (Note 8) is placed in the addition funnel and is added over 5 minutes to the reaction mixture in the round-bottomed flask. This mixture is then transferred to a 250 mL separatory funnel assisted by 60 mL of EtOAc (Note 9), and the aqueous phase is separated (Figure 1E) (Note 10). The organic phase is washed with saturated brine (80 mL) (Note 11) and dried over anhydrous Na2SO4 (20 g) (Note 12). The filtrate is concentrated by rotary evaporation (40 °C, 40 mmHg) and dried under vacuum (23 °C, 5.0 mmHg, 6 h) to give a purple solid (10.5-11.1 g, 56.8-58.1 wt% purity, 64.1-69.3% yield) (Figure 1F), which is used in the next step without further purification (Note 13).

Figure 1. Synthesis of compound 1. A) Reaction setup, B, C) Reaction progress, D) Before quenching, E) Work-up, F) Crude product (photos provided by authors)
A 300 mL, one-necked, round-bottomed flask (24/40 joint) equipped with a 3.5-cm, Teflon-coated magnetic stir bar is charged with 1, THF (40 mL) (Note 14) and MeOH (40 mL) (Note 15) under an air atmosphere. 2 M NaOH (55 mL) (Note 16) is added at 23 °C and the resulting mixture is stirred for 3 h at 23 °C (Note 17) (Figure 2A). The resulting solution (Figure 2B) is concentrated using a rotary evaporator (30 ºC, 30 mmHg) until about ca. 60 mL remains (Figure 2C). The concentrated mixture is transferred to a 300 mL separatory funnel using water (60 mL). The aqueous layer is extracted with Et2O (3 x 80 mL) (Note 18) (Figure 2D). The Et2O layers are discarded. The resulting aqueous layer is transferred to a 300 mL Erlenmeyer flask and Et2O (80 mL) (Note 18) is added and cooled to 0 °C. The resulting mixture is acidified carefully (Note 19) through the addition of 2 M HCl (65 mL) (Note 20) at 0 ºC over a period of 3 min. The resulting mixture is transferred to a 500 mL separatory funnel using Et2O (30 mL). The aqueous layer is separated and extracted with Et2O (100 mL) (Note 18). The combined organic layers are washed with saturated brine (100 mL) (Note 11), dried over anhydrous Na2SO4 (30 g) (Note 12), and filtered through a sintered glass funnel (4 cm diameter, medium porosity) under vacuum suction. The filtrate is concentrated by rotary evaporation (20 °C, 45 mmHg) (Note 21) to give a deep brown oil (Figure 2E). To the resulting oil, containing 3-(4-chloro-1-hydroxynaphthalen-2-yl)propanoic acid (2), is added CHCl3 (50 mL) (Note 22), which is then concentrated by rotary evaporation (20 °C, 45 mmHg) (Note 23) and dried under vacuum (23 °C, 5.0 mmHg) to give a brown solid (Figure 2F). To the resulting solid is added CHCl3 (15 mL) (Note 22) at 0 ºC (Note 24), and the solids are collected by suction filtration on a Büchner funnel (7 cm diameter), and washed with cold (0 ºC) CHCl3 (15 mL) (Note 22) followed by hexane (20 mL) (Note 25). The resulting solid is dried under vacuum (23 °C, 5.0 mmHg, 12 h) to give 2 (3.62-3.98 g, 97.5-99.7 wt%, 36.0-38.7 % yield) as a beige solid (Figures 2G) (Notes 26 and 27).

Figure 2. Synthesis of compound 2. A) Reaction setup, B) Before evaporation, C) After evaporation, D) Work-up, E) Crude, F) Solidification of crude product, G) Product 2 (photos provided by authors)
B. (S)-4'-Chloro-3,4-dihydro-1'H,5H-spiro[furan-2,2'-naphthalene]-1',5-dione (3). An oven-dried 500 mL, three-necked (main 24/40, two sides 15/25 joints), round-bottomed flask is charged with 2 (6.27 g, 25.0 mmol, 1.0 equiv) and N,N'-(2S,2'S)-(2-iodo-1,3-phenylene)bis(oxy)bis(propane-2,1-diyl)bis(2,4,6-trimethylbenzamide (1.61 g, 2.50 mmol, 10 mol%) (Note 28) under an air atmosphere, and the flask is equipped with a 3.5 cm oval Teflon-coated magnetic stir-bar, an internal low-temperature thermometer, an inlet adapter fitted with a nitrogen inlet, and a rubber septum (Figure 3A). A nitrogen line is evacuated under high vacuum (5.0 mmHg, few seconds) and filled with nitrogen (three cycles). The septum is removed, and 1,2-dichloroethane (245 mL) (Note 29) and ethanol (8.76 mL, 150 mmol, 6.0 equiv) (Note 30) are added with a graduated cylinder under nitrogen flow. The septum is replaced. The flask is immersed in an EtOH bath in a low-temperature chamber (Note 31) that is cooled to -20 ºC (Figure 3B) (Note 32). After the internal temperature is reached to -20 ºC, the septum is removed, and meta-chloroperoxybenzoic acid (m-CPBA, 6.72 g, 30.0 mmol, 1.2 equiv) (Note 33) is added slowly (within 1~2 min) via the neck under nitrogen flow. The wall of the neck is washed with 1,2-dichloroethane (5 mL) (Note 29), and the septum is replaced. The reaction mixture is stirred at -20 °C for 24 h under nitrogen atmosphere (Note 34). To the resulting brown solution (Figure 3C) are added saturated aqueous Na2SO3 (50 mL) (Note 35) and saturated aqueous NaHCO3 (50 mL) (Note 36) (Note 37), and then the reaction flask is moved to an ice-bath (0 °C) and stirred for additional 5 min. The resulting mixture is transferred to a 500 mL separating funnel and the aqueous layer is separated (Figure 3D). The organic layer is washed with saturated aqueous NaHCO3 (50 mL) (Note 36)(Figure 3E). The combined aqueous layers are extracted with CHCl3 (2 x 80 mL) (Note 22). The combined organic layers are washed with saturated brine (150 mL) (Note 11), dried over anhydrous Na2SO4 (50 g) (Note 12), and filtered through a sintered glass funnel (6.5 cm diameter, medium porosity) under vacuum suction. The filtrate is concentrated by rotary evaporation (30 °C, 30 mmHg). The crude (Figure 3F) is further purified by column chromatography (Note 38) (Figures 3G-I) to give 3 (5.33 g, 86% yield, 96% ee) (Notes 39, 40 and 41) as a pale-yellow solid (Note 42) (Figure 3J).

Figure 3. Synthesis of compound 3. A) Reaction setup, B, C) Reaction progress, D, E) Work-up, F) Crude product, G) Column chromatography, H) Fractions, I) TLC of fractions, J) Pure product 3 (photos provided by authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
4-chloro-1-naphthol,
toluene,
trifluoromethanesulfonic acid,
acrylic acid,
nitrogen,
sodium hydrogen carbonate,
ethyl acetate,
sodium chloride,
sodium sulfate,
tetrahydrofuran,
methanol,
sodium hydroxide,
diethyl ether,
hydrogen chloride,
chloroform,
hexane,
N,N'-(2S,2'S)-2,2'-(2-iodo-1,3-phenylene)bis(oxy)bis(propane-2,1-diyl)bis(2,4,6-trimethylbenzamide,
1,2-dichloroethane,
ethanol,
meta-chloroperoxybenzoic acid,
sodium sulfite and silica gel, as well as the proper procedures for experimental operations. Although no problems were encountered with the explosion of
m-CPBA or other chemicals in the use of the oxidation procedure (step B), prudence dictates that all operations should be conducted in a hood.
2.
4-Chloro-1-naphthol (≥98%) was purchased from Sigma-Aldrich and used as received.
3. The reaction is performed
under a positive pressure of nitrogen gas by using a
nitrogen-filled
balloon or connecting the reaction setup to nitrogen manifold via a glass adapter on top of the reflux condenser.
4.
Toluene (anhydrous, 99.8%) was purchased from Sigma-Aldrich and used as received.
6.
Acrylic acid (99%, anhydrous) was purchased from Sigma-Aldrich and used as received.
7. The reaction progress could be checked by TLC analysis (TLC Silica gel 60 F254, pre-coated plates (0.25 mm) purchased from Merck) (visualized with 254 nm UV lamp and molybdatophosphoric acid) with
EtOAc/
acetone/
hexane (1:2:10) as an eluent. Product Rf = 0.42, starting material Rf = 0.32. Alternatively, the reaction can be monitored by NMR analysis of the crude reaction mixture: Take 20 µL of crude reaction mixture and dissolve into 600 µL CDCl
3 inside of an NMR tube. If starting material remains, a doublet peak (
J = 8.0 Hz) around 6.75 ppm will appear in
1H NMR spectrum.
8. K
2HPO
4 (≥98%) was purchased from Sigma-Aldrich and used as received. The 25 wt% solution is prepared by dissolving 250 g of salt into 750 mL of deionized water.
9.
EtOAc (≥99.8%) was purchased from Sigma-Aldrich in an amber glass bottom and used as received.
10. This phase separation should be done without extended waiting (<15 min). A flashlight may be needed to assist in observing the separation.
11.
Sodium chloride (
NaCl (≥99.0%)) was purchased from Sigma-Aldrich and used as received. Saturated brined solution was prepared using deionized water.
12.
Sodium sulfate (≥99.0%) was purchased from Sigma-Aldrich and used as received.
13. If desired, compound
1 can be purified on small scale (crude: 107 mg) by flash column chromatography on SiO
2. A column (diameter: 24 mm, height: 500 mm) was charged with 25 g of silica (E. Merck Art. 9385) and
hexane. Sand with 1 cm minimum height (30-50 mesh particle size; purchased from FUJIFILM Wako Pure Chemical Corporation) was added to the top of the column (sand was used to assist packing). The crude residue was transferred to the column. The product was eluted with 0.42 L of
hexane/
EtOAc = 20:1. The product was checked by TLC analysis (TLC Silica gel 60 F254, pre-coated plates (0.25 mm) purchased from Merck) (visualized with 254 nm UV lamp and molybdatophosphoric acid) with
EtOAc/
acetone/
hexane (1:2:10) as an eluent. Product R
f = 0.42. The fractions (18 x 130 mm) 14-20 were collected and concentrated by rotary evaporation (30 °C, 15 mmHg) and dried under vacuum (23 °C, 5.0 mmHg, 6 h). Alternatively, this compound can be purified with an automatic LC purification system (CombiFlash Rf+ from Teledyne ISCO). Mobile phase was ramped from 100% hexanes to 100%
EtOAc over 30 min for the purification. When purified in this way compound
1 was obtained as an off-white solid, which has the following properties: mp 121.5-124.5°C;
1H NMR
pdf (500 MHz, CDCl
3) δ: 2.90 (dd,
J = 8.2, 6.6 Hz, 2H), 3.13 (dd,
J = 8.3, 6.5 Hz, 2H), 7.39 (s, 1H), 7.58-7.65 (m, 2H), 8.20-8.27 (m, 2H).
13C NMR
pdf (125 MHz, CDCl
3) δ: 23.9, 29.1, 117.5, 121.5, 124.6, 124.9, 125.4, 127.3, 127.5, 127.7, 130.6, 146.0, 167.9; IR (film): 2917, 2850, 1754, 1368, 1252, 1172, 1099, 872 cm
-1 Anal. Calcd. For C
13H
9ClO
2: C, 67.11; H, 3.90. Found: C, 67.43; H, 3.83.
14.
THF (≥99.9%) was purchased from Sigma-Aldrich in a Sure/Seal bottle and used as received.
15. MeOH (99.8%) was purchased from Sigma-Aldrich in a Sure/Seal bottle and used as received.
16.
NaOH (≥97.0%) was purchased from Sigma-Aldrich and used as received. 2
M NaOH solution was prepared by dissolving 20 g of solid
NaOH into 250 mL of deionized water.
17. The reaction progress was checked by TLC analysis (TLC Silica gel 60 F254, pre-coated plates (0.25 mm) purchased from Merck) (visualized with 254 nm UV lamp and molybdatophosphoric acid) with
EtOAc/
acetone/
hexane (1:2:10) as an eluent. Product Rf = 0.03, starting material Rf = 0.41.
18. Et
2O (≥99.7%) was purchased from Sigma-Aldrich and used as received.
19. To prevent the acid-catalyzed intramolecular re-lactonization of
2 to
1.
20. Concentrated
HCl (36.5-38.0%) was purchased from Sigma-Aldrich and used as received. 2
M HCl solution was prepared using deionized water.
21. If the concentration was performed at higher temperature (>25 °C),
2 would be converted gradually to lactone
1.
22.
CHCl3 (≥99.8.0%) was purchased from Sigma-Aldrich and used as received.
23.
CHCl3 was used to promote solidification of the crude product. In some cases the residue remains as an oil. Sonication can assist in crashing out the product.
24. To obtain consistent product quality, big chunks of solid at this stage should be broken up with a spatula and the mixture stirred at 0 °C for at least 20 min with magnetic stir bar before filtration.
25.
n-Hexane (≥95.0%) was purchased from Sigma-Aldrich and used as received.
26.
3-(4-Chloro-1-hydroxynaphthalen-2-yl)propanoic acid (
2) has the following properties: mp 107.5 °C (decomposed);
1H NMR
pdf (DMSO-
d6, 500 MHz) δ: 2.57 (t,
J = 7.7 Hz, 2H), 2.99 (t,
J = 7.6 Hz, 2H),7.49 (s, 1H), 7.57 (ddd,
J = 8.1, 6.8, 1.3 Hz, 1H), 7.61 (ddd,
J = 8.1, 6.8, 1.3 Hz, 1H), 8.01-8.06 (m, 1H), 8.25-8.30 (m, 1H), 9.47 (s, 1H), 12.17 (s, 1H);
13C NMR
pdf (DMSO-
d6, 125MHz) δ: 25.0, 33.9, 120.5, 122.5, 122.6, 123.4, 125.7, 126.5, 126.8, 128.4, 129.4, 149.1, 174.1; IR (film): 3362, 3284, 3055, 2944, 2652, 2573, 1689, 1447, 1379, 1235, 880 cm
-1; Anal. Calcd. For C
13H
11ClO
3: C, 62.29; H, 4.42. Found: C, 62.42; H, 4.26.
27. Purity (97.5-99.7 wt%) for the three runs was established by qNMR
pdf using 1,3,5-trimethoxybenzene as an internal standard.
28. Chiral aryliodine catalyst was prepared in an
Org. Synth. procedure
2 that directly precedes this manuscript.
N,N'-[(2S,2'S)-[(2-Iodo-1,3-phenylene)bis-(oxy)]bis(propane-2,1-diyl)]bis(mesitylamide) (Cas No: 1399008-27-5) can be also purchased from Tokyo Chemical Industry Co., Ltd. (TCI).
29.
1,2-Dichloroethane (anhydrous, 99.8%) was purchased from Sigma-Aldrich and used as received.
30.
Ethanol (>99.5%) was purchased from Sigma-Aldrich and used as received.
31. EYELA Low Temp. Pairstirrer PSL-1400 was used.
32. This reaction can also be carried out in a jacketed 500 mL 3-necked, round bottomed flask (all 24/40 joints) connected to a chiller unit. Jacket coolant temperature was set at -24.5 °C to be able to reach -20.0 °C internal reaction temperature.
33.
m-CPBA (77 %) was purchased from Sigma-Aldrich and used as received.
34. The reaction progress was checked by TLC analysis (TLC Silica gel 60 F254, pre-coated plates (0.25 mm) purchased from Merck) (visualized with 254 nm UV lamp and molybdatophosphoric acid) with AcOH/
EtOAc/
hexane (1:50:50) as an eluent. Product R
f = 0.55, starting material R
f = 0.24.
35. Na
2SO
3 (≥98.0%) was purchased from Sigma-Aldrich and used as received. Saturated aqueous Na
2SO
3 solution was prepared using deionized water.
36. NaHCO
3 (≥99.7%) was purchased from Sigma-Aldrich and used as received. Saturated NaHCO
3 solution was prepared using deionized water.
37. Saturated aqueous Na
2SO
3 and NaHCO
3 solutions were pre-cooled in an ice bath (0 °C).
38. The product
3 was purified using column chromatography with a gradient of
EtOAc/
hexane from 20% to 25%. A column (diameter: 55 mm, height: 700 mm) was charged with 130 g of silica (E. Merck Art. 9385) and
hexane. Sand with 2 cm minimum height (30-50 mesh particle size; purchased from FUJIFILM Wako Pure Chemical Corporation) was added to the top of the column (sand was used to assist packing). The crude residue was transferred to the column. The product was eluted with 0.5 L of
hexane/
EtOAc = 4:1 followed by 2.4 L of
hexane/
EtOAc = 3:1. The product was checked by TLC analysis (TLC Silica gel 60 F254, pre-coated plates (0.25 mm) purchased from Merck) (visualized with 254 nm UV lamp and molybdatophosphoric acid) with
EtOAc/
hexane (1:1) as an eluent. Product R
f = 0.53. The fractions (30 x 200 mm) 10-28 were collected and concentrated by rotary evaporation (30 °C, 15 mmHg) and dried under vacuum (23 °C, 5.0 mmHg, 14 h). Alternatively, this compound can be purified with an automatic LC purification system (CombiFlash Rf+ from Teledyne ISCO). Mobile phase was ramped from 100% hexanes to 100%
EtOAc over 30 mins for the purification. The fractions containing product was combined and concentrated by rotary evaporation (30 °C, 100-40 mmHg) and dried under vacuum (23 °C, 5.0 mmHg, 2 h) with N
2 flow on top.
39.
(S)-4'-Chlorospiro[tetrahydrofuran-2,2'-(1'H-naphthaline)]-1',5-dione (
3) has the following properties: mp 101.5-103.6 ºC;
1H NMR
pdf (CDCl
3, 400 MHz) δ: 2.24 (ddd,
J = 13.5, 11.1, 9.7 Hz, 1H), 2.45 (ddd,
J = 13.5, 9.6, 2.3 Hz, 1H), 2.62 (ddd,
J = 17.7, 9.7, 2.3 Hz, 1H), 2.89 (ddd,
J = 17.7, 11.1, 9.6 Hz, 1H), 6.39 (s, 1H), 7.51 (td,
J = 7.5, 1.6 Hz, 1H), 7.72-7.78 (m, 2H), 8.05 (ddd,
J = 7.7, 1.3, 0.6 Hz, 1H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 26.7, 31.6, 83.6, 126.2, 127.5, 128.2, 129.3, 130.2, 131.9, 134.6, 135.9, 175.9, 194.8; IR (film): 3055, 2950, 1780, 1687, 1592, 1291, 1188, 1160, 1025, 941 cm
-1; Anal. Calcd. For C
13H
9ClO
3: C, 62.79; H, 3.65. Found: C, 62.96; H, 3.41.
40. SFC-Conditions: Column: CHIRALPAK IA-3 (3.0 µm, 150 x 4.6 mm) MP A: CO
2; MP B: 25 mM isobutylamine (IBA) in
methanol. Column temperature: 40 °C. Wavelength: PDA. Pressure: 2600 psi. Flow rate: 2.5 mL/min,
Hexane/EtOH = 10:1 as eluent, 1.0 mL/min,
tR = 15.6 min (
R),
tR = 17.4 min (
S); [α]
25.0D = -127.2 (
c 1.00,
CHCl3) for 96% ee;
41. Purity (98.4 wt%) was established by qNMR
pdf using 1,3,5-trimethoxybenzene as an internal standard.
42. A half-scale run was performed, and the product (
3) was obtained 2.91 g (82% yield, 97% purity) with 94% ee.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The enantioselective oxidative dearomatizative coupling of arenols is an useful strategy for the synthesis of various biologically active compounds.
3 The development of enantioselective oxidative dearomatization of arenols using chiral hypervalent iodine catalysis is one of the most challenging areas in asymmetric organocatalysis.
3g,4 Especially, several researchers have pointed out that it is quite difficult to induce asymmetry via a chiral hypervalent iodine reagent because of the exclusive formation of phenoxenium ion via the dissociation of a chiral organoiodine(III) fragment during the reaction (Scheme 1).
3g,5
Scheme 1. General concept of the hypervalent iodine-mediated enantioselective oxidative dearomatizative coupling of arenols
In 2008, Kita and colleagues overcame this difficulty for the first time.
6 They designed a chiral organoiodine with a conformationally rigid 1,1'-spirobiindane backbone, and applied it to the enantioselective oxidative spirolactonization of 1-naphthol derivatives to the corresponding spirolactones with moderate enantioselectivity (up to 69% ee
6a) (Scheme 2).
6 In contrast to Kita's conformationally rigid catalysts, our group introduced the rational design of conformationally flexible hypervalent organoiodines(III) as chiral catalysts based on secondary nonbonding interactions (i.e., intramolecular hydrogen-bonding interactions between the acidic amido protons and the iodine(III) ligands) (Scheme 2).
7,7
Scheme 2. Kita's and our chiral organoiodine catalysts
In 2010, we designed
C2-symmetric organoiodine
1a (1
st-generation catalyst, Ar = Mes) derived from lactate as a chiral source for the catalytic enantioselective oxidative spirolactonization of 1-naphthol derivatives
3 to the corresponding spirolactones
4 with high enantioselectivity of up to 92% ee (Scheme 3).
7
Scheme 3. Enantioselective oxidative spirolactonization of 1-naphthols using 1st-generation catalyst 1a
However, lactate-based
1 was found to be insufficient for the oxidation of phenols
5, which were less reactive than 1-naphthols with respect to not only reactivity but also enantioselectivity.
8a To overcome these limitations, we designed a new chiral iodoarene
2a (2
nd-generation catalyst, Ar = Mes) derived from 2-aminoalcohol instead of lactate as a chiral source (Scheme 4).
8a A catalyst loading of 1 to 10 mol% was enough to give the desired cyclohexadienone spirolactones
6 and the subsequent Diels-Alder adducts
7 with excellent enantioselectivities (up to 99% ee).
Scheme 4. Enantioselective oxidative spirolactonization of phenols 5 using 2nd-generation catalysts 2a
Moreover, we succeeded in rationally controlling the desired associated pathway using alcohol additives such as
methanol or
ethanol for electron-rich phenols (Schemes 4 and 5).
8a On the other hand, the use of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) was crucial to induce high reactivity for the electron-deficient phenols, which seems to disfavor a dissociative path (Scheme 4).
8a,9 This can be explained by the disfavoring of the dissociative path of electron-deficient phenols under these conditions. Furthermore, X-ray diffraction and NOE (Nuclear Overhauser Effect)-NMR analyses of an situ-generated organoiodine(III) showed that a suitable chiral environment around the iodine(III) center was constructed via intramolecular hydrogen-bonding interactions.
7a
Scheme 5. Additional methanol effect on the enantioselective dearomatization of electron-rich phenols
By using our flexible designer organoiodine catalysts
1 or
2, we also achieved the first enantioselective oxidative dearomatization of
ortho- and
para-hydroquinone derivatives
9 or
10 to give the corresponding masked benzoquinones
11 or
12 with high enantioselectivities (Scheme 6).
8b A tether strategy in which phenols were
O-tethered to an acetic acid or
ethanol unit at the
ortho- or
para-position realized rapid intramolecular cyclization enantioselectively prior to dissociation
8a of the chiral iodine moiety. Interestingly, the use of slightly modified catalyst
2b was found to be superior to the use of
2a. Especially, this remote electronic effect was found to enhance enantioselectivity for the oxidation of phenols in a toluene-buffer (
pH 7.0) biphasic solvent. Additionally, to achieve high enantioselectivity for the highly challenging
para-cyclization reaction, we designed new lactate-derived catalysts
1b bearing a deeper chiral cavity.
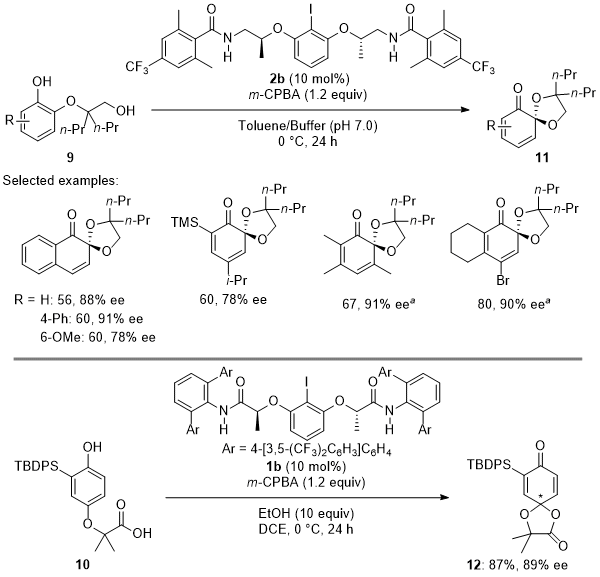
Scheme 6. Enantioselective oxidative spirolactonization of ortho- (9) and para-hydroquinones (10) using designer organoiodine catalysts 2b and 1b, respectively.a 1a was used instead of 1b in CHCl3/H2O
Recently, we also achieved an enantioselective oxidative dearomatization of 2-naphthol derivatives
9 by using our conformationally flexible organoiodine catalyst
2a (Scheme 7).
8c Moreover, excellent enantioselectivities were achieved for 1-naphthol derivatives
3, which had previously
7 been obtained with lower enantioselectivities with lactate-based organoiodine
1.
8c Interestingly, the use of HFIP and
methanol as additives was crucial to induce high enantioselectivity for 2-naphthol and 1-napthols, respectively.
Scheme 7. Enantioselective oxidative spirolactonization of 1- (3) and 2-naphthols (13) using 2nd-generation catalyst 2a. a 2a (10 mol%)
Considering the safety issues and practicality of the large-scale experiments, here, we investigated the reaction conditions and oxidant purity for the oxidative spirolactonization of 1-naphthol
3b (Table 1). To avoid any reproducibility problems, we used purified
m-CPBA (>99%) as an oxidant under diluted conditions (0.02
M) for small scale-reactions.
7,8 Although no problems were encountered with the oxidant during purification, the use of commercially available
m-CPBA (77%) would be preferred in large-scale reactions. Additionally, to make the reaction practical, the use of large amounts of solvents should be avoided in large-scale experiments. First, the oxidation of
3b was performed on 0.5 mmol-scale to give
4b with the same enantioselectivity albeit in slightly lower yield as in the original report,
8c which was performed on 0.1 mmol-scale (entry 2 versus entry 1). We next investigated several parameters in 0.5 mmol-scale reactions and compared the result. To our delight, the same results were obtained with the use of commercially available
m-CPBA (77%) instead of the purified compound (>99%) as an oxidant (entry 3). Next, the amount of solvent used was investigated and 0.1
M was found to the optimal concentration of starting material
3b (entries 4 and 5). Interestingly, the reaction could be performed even at room temperature without any significant loss of enantioselectivity, however, the chemical yield of
4b was decreased due to undesired intramolecular dehydrative lactonization (entries 6-8). Finally, the 50-fold scale-up reaction (25-mmol) under optimized conditions (as in entry 4) could be achieved with almost the same results (entry 9, see procedure section for details).
Table 1. Investigation of the Reaction Parameters for Scale-up
Appendix
Chemical Abstracts Nomenclature (Registry Number)
4-Chloro-1-naphthol: 1-Naphthalenol, 4-chloro-; (604-44-4)
Trifluoromethanesulfonic acid, TfOH: Methanesulfonic acid, 1,1,1-trifluoro-; (1493-13-6)
Acrylic acid: 2-Propenoic acid; (79-10-7)
N,N'-(2S,2'S)-(2-Iodo-1,3-phenylene)bis(oxy)bis(propane-2,1-diyl)bis(2,4,6-trimethylbenzamide: Benzamide, N,N'-[(2-Iodo-1,3-phenylene)bis[oxy[(2S)-2-methyl-2,1-ethanediyl]]]bis[2,4,6-trimethyl-; (1399008-27-5)
meta-Chloroperoxybenzoic acid, m-CPBA: Benzenecarboperoxoic acid, 3-chloro-; (937-14-4)

|
Prof. Muhammet Uyanik was born in Samsun, Turkey, in 1981 and received his Ph.D. from Nagoya University, Japan, in 2007 under the direction of Professor Kazuaki Ishihara. He was appointed as an Assistant Professor at Nagoya University in 2007, and became Associate Professor in 2020. His research focused on the development of halogen-based catalysis for the oxidative transformations. |

|
Shinichi Ishizaki was born in Mie, Japan, in 1996. He received his Bachelor's degree from Nagoya University in 2018. He is continuing his graduate studies at Nagoya University under the supervision of Professors Kazuaki Ishihara and Muhammet Uyanik. His research focused on chiral organoiodine catalysis. |

|
Prof. Kazuaki Ishihara received his Ph.D. from Nagoya University in 1991 under the direction of Professor Hisashi Yamamoto. He had the opportunity to work under the direction of Professor Clayton H. Heathcock at the University of California, Berkeley, for three months in 1988. After completing his postdoctoral studies with Professor E. J. Corey at Harvard University, he joined Professor H. Yamamoto's group at Nagoya University as an Assistant Professor in 1992, and became Associate Professor in 1997. In 2002, he was appointed to his current position as a full Professor. His research interest is the rational design of high-performance catalysts based on acid-base combination chemistry. |

|
Tao Wang joined the Process Research Department of Merck & Co., Inc. in 2018. His research focuses on developing green, sustainable and safe process using state-of-art organic chemistry. Tao obtained his B. S. degree from Peking University in Beijing, China. In 2011, he joined the research group of Thomas Hoye in University of Minnesota to study new benzyne chemistry and develop biomimetic synthesis of natural products. After completion of his Ph.D., Tao joined the laboratory of David MacMillan as a Postdoctoral Scholar at Princeton University. While at Princeton, he explored photoredox-based methodology for small molecules synthesis and pioneered the development of a proximity-based labelling technology in biological systems. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved