Org. Synth. 2021, 98, 171-193
DOI: 10.15227/orgsyn.098.0171
Mild mono-Acylation of 4,5-Diiodoimidazole: Preparation of 1-(5-Iodo-1H-imidazole-4-yl)pent-4-en-1-one
Michael Morgen,
1 Jasmin Lohbeck,
1 and Aubry K. Miller
1*
Checked by Anji Chen, Hari P. R. Mangunuru, Nathaniel D. Kaetzel, Gopal Sirasani, and Chris Senanayake
1. Procedure (Note 1)
A. N-Methoxy-N-methylpent-4-enamide (1). A 1 L round-bottomed flask equipped with a 3.5 cm Teflon-coated barbell-shaped stir bar is loaded with 4-pentenoic acid (25.5 mL, 249.9 mmol, 1.0 equiv) (Note 2) and CH2Cl2 (500 mL) (Note 3). The resulting solution is magnetically stirred (500 rpm) and oxalyl chloride (22.2 mL, 262.3 mmol, 1.05 equiv) (Note 4) is added dropwise at 24 °C via a 50 mL dropping funnel that is capped with a rubber septum (Figure 1A). The addition is complete within 5 min, at which time the dropping funnel is removed and DMF (0.1 mL) (Note 5) is added to the reaction mixture in one portion with a syringe. The solution begins to vigorously bubble.
The reaction flask is quickly connected in series, first to an empty wash bottle and then to a wash bottle filled with a 5 M NaOH solution (200 mL), which itself is vented to the back of the hood (Figure 1B) (Note 6). The colorless reaction mixture is stirred at 24 °C for 2 h, during which bubbling ceases.
Figure 1. (A) Reaction setup consisting of a 1 L flask equipped with a stir bar that is further connected to a dropping funnel - wash bottles are arranged to be quickly connected to the reaction flask. Oxalyl chloride is slowly dropping into a stirring solution of 4-pentenoic acid; (B) Reaction mixture after addition of DMF. The reaction flask is now connected to a series of wash bottles.
(Photos provided by submitters)
The reaction mixture is concentrated on a rotary evaporator (40 °C, 375 then 110 mmHg) leaving the stir bar inside the flask (Note 7). The remaining orange oily residue is dissolved in CH2Cl2 (250 mL) and again concentrated on a rotary evaporator (40 °C, 375 then 110 mmHg) (Note 8). The residue is then dissolved in CH2Cl2 (400 mL), cooled to 0 °C in an ice bath, and N, O-dimethylhydroxylamine hydrochloride (24.35 g, 249.7 mmol, 1.0 equiv) (Note 9) is added in one portion to make a suspension. The flask is equipped with a 100 mL dropping funnel filled with triethylamine (72.7 mL, 524.4 mmol, 2.10 equiv) (Note 10). Triethylamine is added dropwise within 5 to 10 min to the magnetically stirred (500 rpm) reaction mixture at 0 °C (Figure 2A).
After the addition is complete, the reaction mixture is stirred at 0 °C for 2 h and a white suspension forms. The suspension is poured into a 1 L separatory funnel containing a saturated solution of NaHCO3 (400 mL) at 24 °C (Figure 2B). The reaction flask is rinsed with CH2Cl2 (2 x 25 mL) and the resulting solutions are added to the separatory funnel. After shaking, the phases are separated and the yellow-orange organic layer is further washed with water (2 x 300 mL) (Note 11) and a saturated aqueous NaCl solution (300 mL), dried over MgSO4 (50 g) (Note 12), filtered through fluted filter paper in a glass funnel (Note 13), and concentrated on a rotary evaporator (40 °C, 500 then 15 mmHg). The residual orange-colored oil (contaminated with solid Et3N•HCl) is transferred into a 100 mL round-bottomed flask (NS24/40) equipped with a 2.5 cm long oval Teflon coated stir bar (Note 14). This is connected to a short distillation bridge (Liebig condenser) equipped with a thermometer and a distillation cow bearing four 100 mL flasks. The distillation flask is submerged into an oil bath on a magnetic hot plate and is covered with aluminum foil from above the oil level up to the thermometer (Figure 2C). The apparatus is connected via high vacuum-grade silicone tubing to a Schlenk-line and evacuated. By using a needle valve, a pressure of 7-10 mmHg is maintained. After waiting 2-3 min until residual solvent is

Figure 2. (A) Reaction mixture while adding Et3N - a white suspension forms during the addition; (B) Washing of the organic phase with saturated aqueous NaCl; (C) Distillation of the product using a Liebig condenser equipped with a thermometer and a distillation cow bearing four flasks; (D) Appearance of pure 1.
(Photos provided by submitters)
removed (indicated by ceased bubble formation) the oil bath temperature is gradually increased from 24 °C to 95 °C. The pure product distills at a temperature of 85 °C (oil bath temperature: 95 °C) and a pressure of 7-10 mmHg (Note 15). Weinreb amide 1 is obtained as a colorless oil in a yield of 69% (24.69 g, 172.4 mmol) (Figure 2D) (Note 16 and 17).
B. 4,5-Diiodo-1H-imidazole (2). A 500 mL Erlenmeyer flask equipped with a 4.0 cm long Teflon-coated flat stir bar is charged with potassium iodide (97.53 g, 587.6 mmol, 4.0 equiv) (Note 18). Water (300 mL) (Note 11) at 24 °C is then added to make a colorless solution. Iodine (74.56 g, 293.8 mmol, 2.0 equiv) (Note 19) is then added and the resulting mixture is magnetically stirred (700 rpm) until complete dissolution of iodine (~15-20 min) (Note 20) provides a dark brown solution.
In parallel, a 1 L round-bottomed flask equipped with a 3.2 cm long oval Teflon-coated stir bar is charged with a 2 M NaOH solution (NaOH (35.25 g, 881.3 mmol, 6.0 equiv) in H2O (440 mL)) (Note 21). To this is added imidazole (10.0 g, 146.9 mmol, 1.0 equiv) (Note 22) at 24 °C to make a colorless solution. This flask is connected to a 500 mL dropping funnel capped with a rubber septum, which is filled with the before mentioned KI/I2 solution (Figure 3) (Note 23).
Figure 3. Reaction setup before the addition of the KI/I2 solution.
(Photo provided by submitters)
The KI/I2 solution is added dropwise to the imidazole solution at 24 °C within 60-90 min. Upon addition, a brownish color with concomitant formation of a white precipitate is observed, both of which rapidly disappear (Figure 4A). After complete addition, the resulting yellow solution (Figure 4B) is stirred at 24 °C for 3 h, during which the solution becomes pale-yellow to colorless (Figure 4C). The reaction mixture is cooled to 0 °C using an ice bath and acidified. This is done by adding concentrated HCl (~48-49 mL, ~4 equiv) (Note 24) portion wise (5 x 9 mL) and then dropwise to the reaction mixture, during which a number of visual changes are observed. After each of the five 9 mL additions, a dark red/brown precipitate forms, which slowly re-dissolves with vigorous stirring (Figure 4D). The solution remains essentially colorless. As the endpoint nears, dropwise addition of HCl produces a white precipitate and a yellow/orange color in the solution, both of which disappear with stirring. Closer to the endpoint, the white precipitate persists (Figure 4E). The endpoint can be determined by color change when a yellow/orange color of the solution no longer returns to colorless. Temporarily discontinuing stirring can be helpful for making this determination. The resulting mixture is pH 8, which can be checked by pH-indicator paper. The resulting white solid is collected by vacuum filtration using a round filter paper (ø = 9 cm) in a Büchner funnel and washed with cold (4 °C) water (2 x 250 mL). The solid material is air dried in the funnel for 1 h with house vacuum (Figure 4F) and transferred into a 1 L round-bottomed flask, using EtOH (Note 25) to quantitate the transfer. The resulting suspension is heated to reflux on a boiling water bath (Note 26) with EtOH (350 mL) to fully dissolve the solids (Note 27). The orange-colored solution is allowed to cool to 24 °C on the benchtop (Figure 4G) and afterwards in the freezer at -20 °C for 18 h. The resulting solid is collected by vacuum filtration on filter paper (ø = 9 cm) in a Büchner funnel and washed with cold (-20 °C) ethanol (2 x 50 mL) and air dried on the filter for 1 h using house vacuum. The product is obtained as an off-white solid. The mother liquor is concentrated to dryness on a rotary evaporator (40 °C, 35 mmHg), suspended in 250 mL of EtOH and recrystallized as described above to obtain a second crop of 2. The two crops are combined, finely mortared, transferred to a 250 mL round-bottomed flask (NS 24/40), and dried under high vacuum (24 °C, 2 x 10-2 mmHg) for 18 h to give 4,5-diiodoimidazole (2) as an off-white solid in a yield of 59% (27.90 g, 87.22 mmol) (Figure 4H) (Note 28 and 29).

Figure 4. (A) Reaction mixture while adding the KI/I2 solution; (B) Immediately after complete addition of the KI/I2 solution; (C) After stirring at 24 °C for 3 h; (D) While acidifying with conc. HCl; (E) After adjusting to pH 8 and continued stirring for 2 min; (F) Material obtained after vacuum filtration of the precipitated product; (G) Dissolved in hot ethanol for recrystallization; (H) Final product 2.
(Photos provided by submitters)
C. 1-(5-Iodo-1H-imidazole-4-yl)pent-4-en-1-one (3). An oven-dried 1 L three-necked, round-bottomed flask (3 x NS 24/40) is filled with anhydrous THF (500 mL) (Note 30 and 31), equipped with two glass stoppers, and connected to a Schlenk line with a glass adapter. Under positive nitrogen pressure, the middle glass stopper is exchanged with a glass rod equipped with a 5 cm long oval-shaped Teflon vane that is connected to an overhead stirrer (Figure 5A). Via the remaining side neck, lithium chloride (3.31 g, 78.2 mmol, 1.0 equiv) (Note 32) is added and the mixture is stirred (160 rpm) (Note 33) at 24 °C under an atmosphere of nitrogen until the lithium chloride is fully dissolved (~18 h). Then finely crushed 4,5-diiodoimidazole 2 (25.0 g, 78.2 mmol, 1.0 equiv) (Note 34) is added. The resulting yellow solution is cooled to 0 °C using an ice bath, whereupon a white precipitate forms (Note 35). Then the glass stopper is replaced with a silicone septum and methylmagnesium chloride (3 M solution in THF; 27.4 mL, 82.1 mmol, 1.05 equiv) (Note 36 and 37) is added dropwise with a syringe to the reaction mixture within 5-10 min (Note 38). During addition of methylmagensium chloride the reaction mixture clarifies, and the resulting orange solution is stirred (160 rpm) at 0 °C. After 5 min the ice bath is removed and stirring is continued for another 5 min (Note 39) (Figure 5D). Then isopropylmagnesium chloride lithium chloride complex (1.3 M solution in THF; 66.1 mL, 86.0 mmol, 1.10 equiv) (Note 40 and 41) is added with a syringe to the reaction mixture within 10-15 min. The resulting off-white suspension is stirred (160 rpm) (Note 33) at 24 °C for 2.0-2.5 h until the iodine/magnesium exchange is considered complete by TLC monitoring (Note 42) (Figure 5E). Afterwards, neat Weinreb amide 1 (12.31 g, 86.0 mmol, 1.10 equiv) is added to the suspension dropwise with a syringe within 2-3 min (Note 43) and stirring (160 rpm) is continued for 2.0-2.5 h at 24 °C, during which the reaction mixture clarifies again to an orange-colored solution (Figure 5F). The reaction is monitored by TLC (Note 44). After completion, the reaction mixture is transferred into a 2 L separatory funnel and quenched at 24 °C by adding a saturated aqueous solution of NH4Cl (400 mL). The reaction flask is rinsed with saturated aqueous NH4Cl (25 mL) and EtOAc (25 mL) and the resulting mixture is added to the separatory funnel. The phases are separated and the aqueous layer is extracted with EtOAc (2 x 200 mL) (Note 45). The combined organic phases are washed with brine (400 mL), dried with MgSO4 (50 g) (Note 12), filtered through a fluted filter paper in a glass funnel (Note 46), and concentrated on a rotary evaporator (40 °C, 35 mmHg). The obtained off-white solid is crystallized as follows: on a gently boiling water bath, the solid is dissolved in 100 mL of EtOAc (Note 26). To the boiling hot solution is added in a portionwise fashion enough hexanes (~50 mL) (Note 47) so that the white precipitate that forms upon addition slowly disappears. The flask is then covered with a plastic yellow cap and allowed to cool to 24 °C on the bench and then at -20 °C in a freezer for 18 h. The resulting solid is collected by vacuum filtration through a round filter paper (ø = 9 cm) in a Büchner funnel, successively washed with a 1:1-mixture of cold (4 °C) EtOAc/hexanes (100 mL), cold (4 °C) hexanes (100 mL), and afterwards air dried with suction for 30 min (Note 48). The material is further dried under high vacuum (24 °C, 2 x 10-2 mmHg) for 18 h. The desired product 3 is obtained in a yield of 78% (16.91 g, 61.25 mmol) (Figure 5G) (Note 49 and 29).

Figure 5. (A) Reaction apparatus consisting of a 1 L three-necked flask, which is connected to a Schlenk line (positive nitrogen pressure) via one side neck, to an overhear stirrer via the middle neck, with the remaining side neck equipped with a glass stopper; (B) Appearance of the reaction mixture containing LiCl in THF immediately after adding 4,5-diiodoimidazole (2); (C) Upon cooling to 0 °C in an ice bath - an off-white suspension forms; (D) Appearance of the reaction mixture after complete addition of MeMgCl - the suspension clarifies; (E) Appearance of the reaction mixture upon addition of i-PrMgCl•LiCl - again a white suspension is obtained; (F) Appearance of the reaction mixture upon complete addition of the Weinreb amide 1 - an orange-colored solution is obtained; (G) Appearance of the pure product 3 after work-up, recrystallization from hot EtOAc and hexanes and drying under high vacuum.
(Photos provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
4-pentenoic acid,
dichloromethane,
oxalyl chloride,
dimethylformamide,
N, O-dimethylhydroxylamine hydrochloride,
triethylamine,
potassium iodide,
iodine,
imidazole,
sodium hydroxide,
hydrochloric acid,
ethanol,
tetrahydrofuran,
lithium chloride,
methylmagnesium chloride,
isopropylmagnesium chloride,
ethyl acetate and
hexanes, as well as the proper procedures for vacuum distillations.
2.
4-Pentenoic acid (97%) was obtained from Sigma-Aldrich and was used as received.
3.
Dichloromethane (≥ 99.8%, stabilized with 40-150 ppm amylene, anhydrous) was obtained from Sigma-Aldrich and was used as received.
4.
Oxalyl chloride (≥ 99%) was obtained from Sigma-Aldrich and was used as received.
5.
Dimethyl formamide (99.8%, anhydrous) was obtained from Sigma-Aldrich and was used as received.
6. This reduces the copious amount of evolving
HCl from being released into the hood. CO
2 and CO are also produced as gaseous byproducts in this reaction.
7. The product (4-pentenoyl chloride) has a reported boiling point of 125 °C. Care must be taken not to co-distill the product on the rotary evaporator. The product also has an unpleasant odor and it is advisable to use a rotary evaporator that is housed in a ventilated hood, and transport the flask through the lab only while capped.
8. Redissolving the residue and subsequent concentration ensures the removal of any unreacted
oxalyl chloride and residual
HCl.
9.
N, O-Dimethylhydroxylamine hydrochloride (98%) was obtained from Sigma-Aldrich and was used as received.
10.
Triethylamine (≥ 99.5%) was obtained from Sigma-Aldrich and was used as received.
11. Deionized water was used from in-house supply.
12.
Magnesium sulfate (>99.5%, anhydrous) was obtained from Sigma Aldrich and was used as received.
13. The remaining
MgSO4 was washed once with 20 mL of
CH2Cl2.
14. The reaction flask is rinsed with
CH2Cl2 (2 x 2 mL) to quantitate the transfer. The residual solvent was removed on a rotary evaporator (40 °C, 15 mmHg) prior to distillation.
15. A small forerun of a few drops was taken before collecting the pure product
1.
16. A second run on the same scale provided 24.37 g (68%) of the product. The following analytical data were obtained for
1: R
f = 0.5 (
EtOAc/
hexanes 1:1); bp 85 °C (7-10 mmHg);
1H NMR
pdf (600 MHz, CDCl
3) δ: 2.35 - 2.41 (m, 2H), 2.48 - 2.55 (m, 2H), 3.17 (s, 3H), 3.67 (s, 3H), 4.98 (ddt,
J = 10.2, 2.0, 1.4 Hz, 1H), 5.06 (dq,
J = 17.1, 1.6 Hz, 1H), 5.85 (ddt,
J = 16.8, 10.2, 6.5 Hz, 1H).
13C NMR
pdf (150 MHz, CDCl
3) δ: 28.5, 31.2, 32.1, 61.2, 115.2, 137.5, 173.9. LCMS-ESI (
m/z): [M + H]
+ calcd for C
7H
14NO
2+: 144; Found: 144. The purity was determined to be 98.6% by qNMR
pdf spectroscopy in CDCl
3 using 33.3 mg of compound (
1) and 5.3 mg of dimethyl fumarate (99%) as an internal standard.
17. After one month storage at 4 °C in a refrigerator, the submitters observed no decomposition of
1.
18.
Potassium iodide (99%, ACS reagent) was obtained from Sigma-Aldrich and was used as received.
19.
Iodine (99.8%, ACS grade) was obtained from VWR and was used as received.
20. Occasionally the beaker was placed into an ultrasonication bath and small pieces of
iodine had to be broken up with a spatula to facilitate dissolution.
21.
NaOH (97%, pellets) was obtained from Oakwood Chemical and was used as received.
22.
Imidazole (99%) was obtained from Sigma-Aldrich and was used as received.
23.
Potassium iodide was used in this reaction to increase the solubility of
iodine in water by forming potassium triiodide (KI
3).
24.
Hydrochloric acid (36.5-38%, ACS grade) was obtained from Oakwood Chemical and was used as received.
25.
Ethanol absolute (200 proof) was obtained from KOPTEC (analytical reagent grade) and was used as received.
26. A wooden stick was placed in the flask to prevent bumping.
27. In the case that small residual particles did not easily go into solution the solid was gently crushed with a spatula and the flask was put into an ultrasonic bath for half a minute.
28. A second run on the same scale provided 27.73 g (59%) of the product. The following analytical data were obtained for
2: R
f = 0.46 (
CH2Cl2/
MeOH 9:1 + 0.5% NH
4OH). mp (uncorrected): 201-202 °C (provided by submitters).
1H NMR
pdf (600 MHz,
d6-DMSO) δ: 7.78 (s, 1H), 12.91 (br s, 1H).
13C NMR
pdf (150 MHz,
d6-DMSO) δ: 78.5, 95.8, 141.6. LCMS-ESI (
m/z): [M + H]
+ calcd for C
3H
3I
2N
2+: 321, Found: 321. The purity was determined to be 99.6% by qNMR
pdf spectroscopy in
d6-DMSO using 29.2 mg of compound (
2) and 13.3 mg of dimethyl fumarate (99%) as an internal standard.
29. The compound was stored at 4 °C in the dark (with aluminum foil covering the storage bottle).
30.
Tetrahydrofuran (
THF, anhydrous, ≥99.9%, inhibitor-free) was obtained from Sigma-Aldrich.
31. The submitters obtained
tetrahydrofuran (
THF, Chromosolv Plus for HPLC, inhibitor-free, ≥99.9%) from Honeywell Riedel-de Haën and dispensed it from a solvent purification system (MB SPS-800, MBraun) under an atmosphere of nitrogen. The submitters proceeded as follows to obtain the exact amount of solvent: The desired reaction flask was filled with 500 mL of
THF, which had been accurately measured with a graduated cylinder. The solvent level in the flask was labelled with a permanent marker. This filling mark was then used to measure the correct amount of solvent (after oven drying the flask) from the solvent purification system.
32.
Lithium chloride was obtained from Sigma-Aldrich (≥99%, ACS reagent) and was used as received.
33. The checkers used a IKA EUROSTAR 60 digital overhead stirrer, which was set on 160 rpm during the reaction mixture stirring.
34.
4,5-Diiodoimidazole (
2) is now commercially available from a number of suppliers. An attempt to use commercial
2 (ABCR, Karlsruhe, Germany) in this reaction resulted in no conversion. Recrystallization of purchased
2 as described above resulted in material that performed as well as the "home-made"
2. The submitters hypothesize the commercial material was wet.
35. Solutions of
4,5-diiodoimidazole 2 and
LiCl in dry
THF, regardless of temperature or concentration, form thick white precipitates within minutes (see discussion section). At larger reaction scales, the mixture cannot be stirred magnetically, and overhead stirring is required.
36.
Methylmagnesium chloride (3 M solution in
THF) was obtained from Sigma-Aldrich and was used as received.
37. The checkers used a 50 mL plastic syringe with a 4 inch long, gauge 22 needle to measure the amount of
methylmagnesium chloride solution used.
38. Significant bubbling was observed, resulting from methane which was formed by deprotonation of
4,5-diiodoimidazole (
2) with
methylmagnesium chloride.
39. In case of remaining material sticking on the wall of the flask, the stirring rate was temporarily increased in order to wash that material down into the reaction mixture.
40.
Isopropylmagnesium chloride lithium chloride complex solution (1.3 M in
THF) was obtained from Sigma-Aldrich and was used as received.
41. The checker used two 50 mL plastic syringe with a 4 inches long gauge 22 needle to measure the amount of
isopropylmagnesium chloride lithium chloride complex solution used.
42. For reaction monitoring thin-layer chromatography with silica gel 60 F
254-plates from Merck, Darmstadt, Germany and a mixture of
CH2Cl2/
MeOH 9:1 (+ 0.5% NH
4OH) as eluent was used (Figure 6). A mini-work-up of the reaction mixture was performed by removing the septum, pulling ~0.1 mL of reaction mixture into a glass pipette, and quenching this into ~1.0 mL of saturated aqueous ammonium chloride.
EtOAc (~0.2 mL) was added, and after agitation and separation of the two layers, the top organic layer was used for TLC spotting. During this work-up, the desired Grignard intermediate is converted to
4-iodoimidazole, which is what is observed as "product" on the TLC plate, showing an R
f value of 0.40 in
CH2Cl2/
MeOH 9:1 (+ 0.5% NH
4OH) under UV light and with
iodine-staining. The starting material is visible under UV light and shows an R
f value of 0.46 in
CH2Cl2/
MeOH 9:1 (+ 0.5% NH
4OH). The
magnesium/
iodine exchange reaction does not always go to completion, but the next step can be conducted with an observed outcome as shown below. The submitters found that prolonged stirring or a larger excess of
i-PrMgCl•LiCl led to undesired byproducts (see discussion section).
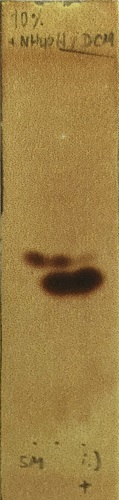
Figure 6. Reaction monitoring of the magnesium/halogen exchange reaction of 4,5-diiodoimidazole (2) with i-PrMgCl•LiCl (left lane: 4,5-diiodomidazole (2); right lane: reaction mixture quenched with conc. NH4Cl solution and extracted with EtOAc; middle lane: co-spot). (Photo provided by submitters)
43. The checkers weighed
1 directly into a tared 12 mL plastic syringe with a 4 inches long gauge 22 needle. After complete addition of
1, the transfer was quantitated by two times pulling 1 mL of reaction mixture into the syringe and dispensing it back into the reaction mixture.
44. For reaction monitoring thin-layer chromatography with silica gel 60 F
254-plates from Merck, Darmstadt, Germany and a mixture of
CH2Cl2/
MeOH 9:1 (+ 0.5% NH
4OH) as eluent was used (Figure 7). A mini work-up was performed as in
Note 42. This was spotted in comparison to the previous quench (
4-iodoimidazole) as well as Weinreb amide
1. The desired product was visible under UV light and stained with
iodine showing an R
f value of 0.50.
Figure 7. Reaction monitoring of the Grignard reaction with Weinreb amide 1 using iodine staining (first lane from left: "4-iodoimidazole" (see above); third lane: reaction mixture; fifth lane: Weinreb amide 1; second and fourth lanes: co-spots). Traces of starting material (2) show-up after long staining time; the reaction can be worked-up with an observed outcome as shown above. (Photo provided by submitters)
45.
Ethyl acetate was obtained from Oakwood Chemical (99.9%, ACS grade) and was used as received.
46. The remaining
MgSO4 was washed once with 20 mL of
ethyl acetate.
47.
Hexanes was obtained from Oakwood Chemical (45%
n-hexane ACS grade) and was used as received.
48. A second and third crop of material was obtained by concentrating the mother liquor and recrystallizing the residue from a smaller volume of
ethyl acetate and
hexanes as mentioned above.
49. A second run on the same scale provided 16.77 g (78%) of the product. The following analytical data were obtained for compound
3: R
f = 0.50 (
CH2Cl2/
MeOH 9:1 + 0.5% NH
4OH).
1H NMR
pdf (600 MHz,
d6-DMSO) δ: 2.30 - 2.37 (m, 2H), 3.01 (t,
J = 7.4 Hz, 2H), 4.97 (d,
J = 10.1, 1H), 5.05 (d,
J = 17.2, 1H), 5.80 - 5.91 (m, 1H), 7.88 (s, 1H), 13.25 (br s, 1H).
13C NMR
pdf (150 MHz,
d6-DMSO) δ: 27.6, 38.1, 115.1, 137.6, 140.6 (see printed spectra for more detail). LCMS-ESI (
m/z): [M + H]
+ calcd for C
8H
10IN
2O
+: 277, Found: 277. The purity was determined to be 98.6% by qNMR
pdf spectroscopy in CDCl
3 using 10.4 mg of compound (
3) and 7.4 mg of dimethyl fumarate (99%) as an internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Imidazole is an important and commonly used heterocycle, finding uses in a wide range of chemistry-related disciplines. Imidazoles are precursors for 1,3-dialkyl imidazolium salts,
2 which are used as ionic liquids and as precursors for the synthesis of
N-heterocyclic carbenes (NHCs) and their metal complexes.
3 In addition, imidazoles are found in many drugs and pesticides as exemplified by the H
2-receptor antagonist Cimetidine and the AT
1-receptor antagonist Losartan as well as Cyazofamid, all of which are 4,5-substituted imidazoles (Figure 8). Thus, the development of reliable and scalable procedures for the regioselective synthesis of sophisticated 4,5-disubstituted imidazoles remains a highly valuable task.
Figure 8. Examples of imidazole-containing compounds.
Herein, we report a multigram-scale synthesis of
1-(5-Iodo-1H-imidazole-4-yl)pent-4-en-1-one (
3) that arose as an important intermediate in a drug discovery project focusing on irreversible methionine aminopeptidase 2 (MetAP2) inhibitors. Starting from
3,
N-alkylation, Heck reaction, and epoxidation gave a library of epoxyimidazoles as our first-generation MetAP2 inhibitors (Figure 9).
4 These substances were designed to mimic the natural product ovalicin.
Figure 9. Synthetic route from 3 to putative MetAP2 inhibitors.
Our first reported synthesis of
34 also called for magnesiation of
4,5-diiodoimidazole (
2) using conditions initially described by Kopp and Knochel.
5 In that case, the resulting Grignard intermediate was treated with 4-pentenal to give an alcohol, which was oxidized to
3 with IBX (Scheme 1). Although this sequence reliably gave good yields on scale, it had drawbacks. First, 4-pentenal is low-boiling (bp 96 °C) and has an extremely noxious odor, making its large-scale laboratory preparation unpleasant.
6 Second, using a reagent at the aldehyde oxidation state necessitated an oxidation to arrive at
3. We therefore developed the current route which gives
3 directly from Weinreb amide
1, which itself is prepared in a two-step one pot reaction from inexpensive
4-pentenoic acid.
7
Scheme 1. Original (lower) and current (upper) approaches to building block 3.
It is of interest to note that at the outset of our MetAP2 inhibitor project (2009) multiple publications reported that reaction of
imidazole with ≥3 equivalents of
I2 produced triiodoimidazole.
8 This apparent error can be traced back to 1908 when Pauly
et al. first described the iodination of different
N-heterocycles, including
imidazole.
9 Due to the limited analytical techniques available at the time, only melting point and elemental analysis were used to characterize the product. The authors wrote of extreme difficulties in obtaining an accurate elemental analysis, which is assuredly the source of the error. Interestingly, this error apparently was not uncovered, or at least not reported in the primary literature, for nearly 100 years. While our preparation only calls for 2 equivalents of
I2, we have found that additional equivalents do not promote further iodination. This has been corroborated in the meantime by others.
10As mentioned above,
THF solutions of
2 and
LiCl produce a white precipitate, which on large scale can cause problems with stirring. Curious to determine what adduct was formed, we prepared a dilute
THF solution of equimolar
2 and
LiCl and stored it at 4 °C. Thin colorless needles formed, which were amenable to X-ray crystallographic analysis. The experimental structure shows a tetrahedral complex of lithium, which is coordinated by two
THF solvent molecules, chlorine and N3 of
4,5-diiodoimidazole (
2) (Figure 10, left). In the solid state, this complex forms a network through intermolecular H-bonds of N
1H and chlorine (Figure 10, right).
11
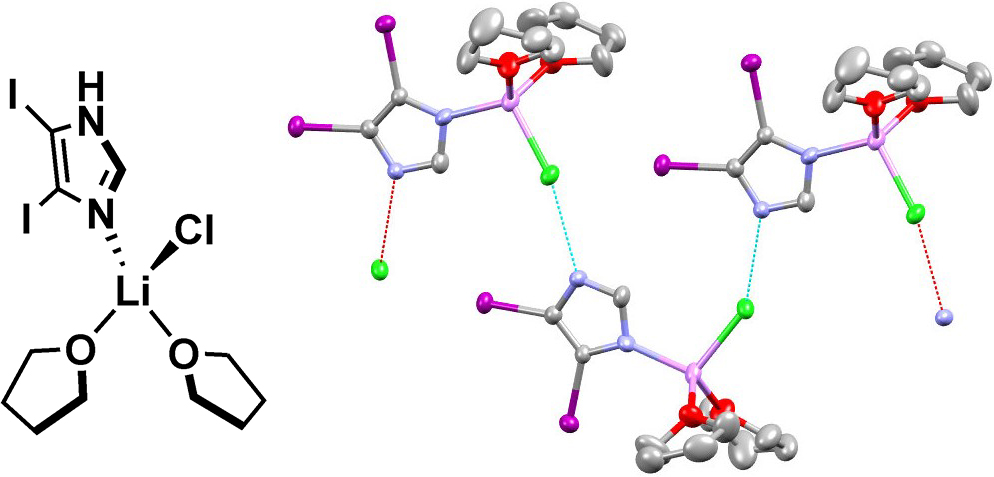
Figure 10: (Left) Depiction of the precipitated adduct of 4,5-diiodoimidazole (2) and LiCl in THF; (Right) the experimental structure of the tetrahedral complex. Ellipsoids are shown at 50% probability level; color code: carbon, grey; oxygen, red; nitrogen, blue; iodine, purple; lithium, pink; chlorine, green; hydrogen omitted for clarity; dashed lines indicate the network formed by this complex via H-bonds (imidazole-N1H--Cl).
As described above, we often observed that
magnesium/
iodine exchange of
2 with 1.1 equiv of
i-PrMgCl•LiCl did not go to completion (TLC monitoring of a mini work-up showed small amounts of remaining
4,5-diiodoimidazole (
2)). An attempt to drive the reaction to completion with additional
i-PrMgCl followed by a prolonged reaction time with
1 (over night; approximately 16 h) led to a byproduct via side reaction with
THF which we identified as 5-iodo-4-(tetrahydrofuran-2-yl)-1
H-imidazole (Scheme 2). The compound is highly crystalline, difficult to remove from the desired product, and has an R
f value (in 9:1
MeOH/CH
2Cl
2 + 0.5% NH
4OH) that is identical to
4-iodoimidazole. The reaction of aryl Grignards with
THF, mediated by alkyl Grignards and iodoalkanes, at room temperature has been reported.
12 In our system, there is an aryl Grignard (the desired
magnesium/
iodine exchange product), an alkyl Grignard (excess
i-PrMgCl), an iodoalkane (
i-PrI - the other
magnesium/
iodine exchange product), and
THF as solvent.
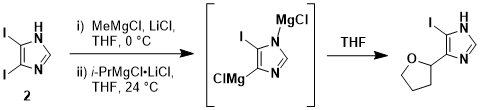
Scheme 2. Observed side-product of the iodine-magnesium exchange reaction in THF as solvent (at a prolonged reaction time).
Appendix
Chemical Abstracts Nomenclature (Registry Number)
4-Pentenoic acid; (591-80-0)
Oxalyl chloride: Ethanedioyl dichloride; (79-37-8)
N, O-Dimethylhydroxylamine hydrochloride: Methanamine, N-methoxy-, hydrochloride (1:1); (6638-79-5)
Triethyl amine: Ethanamine, N,N-diethyl-; (121-44-8)
4,5-Diiodoimidazole: 1H-Imidazole, 4,5-diiodo-; (2) (15813-09-9)
Potassium iodide; (7681-11-0)
Iodine; (7553-56-2)
Imidazole: 1H-Imidazole; (288-32-4)
Sodium hydroxide; (1310-73-2)
Hydrochloric acid; (7647-01-0)
Lithium chloride; (7447-41-8)
Methylmagnesium chloride (3M solution in THF); Magnesium, chloromethyl- (676-58-4)
iso-Propylmagnesium chloride lithium chloride complex: Magnesate(1-), dichloro(1-methylethyl)-, lithium (1:1); (745038-86-2)
1-(5-Iodo-1H-imidazol-4-yl)pent-4-en-1-one: 4-Penten-1-one, 1-(5-iodo-1H-imidazol-4-yl)-; (3) (1585257-59-5)

|
Michael Morgen received his Ph.D. in inorganic chemistry from the Ruprecht-Karls-University, Heidelberg in 2013 while working with Prof. Dr. Peter Comba on the design of novel bispidine-based multimodal imaging agents. Afterwards he joined the Cancer Drug Development Group of Dr. Aubry Miller and Dr. Nikolas Gunkel at the German Cancer Research Center (DKFZ). His current research interests lie in the design and synthesis of organic and metal-organic inhibitors for cancer-related drug targets. |

|
Jasmin Lohbeck was trained as chemistry laboratory assistant at BASF in Ludwigshafen, Germany, from 2006 - 2009. She subsequently joined the chemistry lab of Dr. Aubry Miller at the German Cancer Research Center and specialized in preparative organic synthesis.
|

|
Aubry Miller leads the Cancer Drug Development Group at the German Cancer Research Center (DKFZ) in Heidelberg, Germany. His laboratory focuses on the discovery and biological characterization of chemical probes for use as tools and drug leads in cancer research. |

|
Dr. Anji Chen received his Ph.D. in organic chemistry at Old Dominion University, VA under the mentorship of Prof. Guijun Wang in 2018, where he made contributions on two distinct areas of research: 1) synthesis of new classes of glycoclusters and glycomimetics and their applications as soft biomaterials and 2) synthesis of novel bis-triazole linked carbohydrate-based macrocycles and their application for accelerating copper sulfate mediated click reaction. After completing his graduate studies in 2019, Anji joined TCG GreenChem, Inc. as a post-doctoral researcher. His research interests include the design and development of green and robust large-scale production of active pharmaceutical ingredients (APIs) and synthetic methodology of novel organic transformations. |

|
Hari P. R. Mangunuru earned his Ph.D. in organic chemistry from the Old Dominion University, 2012 under the guidance of Prof. Guijun Wang, where his research focused on developing carbohydrate based hydrogelators as drug delivery carriers. He joined the Department of Process Research & Development at Boehringer Ingelheim, Ridgefield, CT, USA, as a postdoctoral fellow, where he developed a new class of phosphine ligands for asymmetric catalysis. In 2016 he moved to Virginia Commonwealth University as a postdoctoral fellow to work with Prof. B. Frank Gupton, where he was responsible for the discovery and development of green, robust, and safe synthetic routes to active pharmaceutical ingredients (APIs) using continuous flow technology. Currently working as a Senior scientist in Process Chemistry at TCG GreenChem, Inc., his research interests include the development, study, applications of novel organic transformations and continuous flow asymmetric catalysis. |

|
Nathaniel Kaetzel received his B.S. in chemistry from Virginia Commonwealth University while under the mentorship of Prof. Joshua Sieber, where he studied novel total synthetic applications of copper-catalyzed reductive coupling reactions, and Prof. Christopher Kelly, where he studied the oxidative formation of nitriles utilizing an oxoammonium salt, as well as the reactivity of silylated bis-boronic ester species. After completing his undergraduate studies in 2020, Nathaniel joined TCG GreenChem Inc. as a research scientist. His research interests include the instrumental analysis of in-development manufacturing processes for active pharmaceutical ingredients (APIs), as well as the design and synthesis of new chiral phosphine ligands. |

|
Dr. Gopal Sirasani received his Bachelor's and Master's degrees in Hyderabad, India. He obtained his Ph.D. in synthetic organic chemistry in 2011 from Temple University, Philadelphia under the guidance of Prof. Rodrigo B. Andrade. His doctoral research was focused on developing novel methodologies, total syntheses of natural products and their analogs thereof. He got his post-doctoral training in the laboratory of Prof. Emily Balskus at Harvard University, where he developed biocompatible organic reactions utilizing microbially generated reagents to realize transition metal catalysis in the presence of microbes. In 2013, Gopal began his industrial career at Melinta Therapeutics, NewHaven, CT. He is currently working at TCG GreenChem, Inc. as a Director in the department of process research and development. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved