Org. Synth. 2021, 98, 315-342
DOI: 10.15227/orgsyn.098.0315
Preparation of 1-Benzyl-7-methylene-1,5,6,7-tetrahydro-4H-benzo[d]imidazol-4-one
Submitted by Michael Morgen, Jasmin Lohbeck, and Aubry K. Miller*
1Checked by Nageswara Rao Kalikinidi, Venumadhav Janganati, Gopal Sirasani, and Chris Senanayake
1. Procedure (Note 1)
A. 1-(1-Benzyl-5-iodo-1H-imidazol-4-yl)pent-4-en-1-one (2). A 250 mL round-bottomed flask equipped with a 3.2 cm long oval stir bar is charged with 1-(5-iodo-1H-imidazol-4-yl)pent-4-en-1-one (1) (14.00 g, 50.71 mmol, 1.0 equiv) (Note 2) followed by DMF (100 mL) (Note 3). The flask is capped with a rubber septum and the resulting, orange-colored solution is magnetically stirred (500 rpm) and cooled to 0 °C using an ice bath. Potassium carbonate (10.51 g, 76.06 mmol, 1.5 equiv) (Note 4) is added in one portion followed by the addition of benzyl bromide (6.3 mL, 53.2 mmol, 1.05 equiv) (Note 5) dropwise using a syringe over a time period of 2 min. The resulting suspension is stirred at 0 °C for 2 h (Figure 1A). Analysis by TLC (Note 6) is performed to ensure that starting material is consumed. After 1 is entirely consumed the reaction mixture is transferred to a 1 L separatory funnel. The reaction vessel is sequentially rinsed with H2O (50 mL) (Note 7) and EtOAc (50 mL) (Note 8), both of which are transferred to the separatory funnel. Additional H2O (250 mL) and EtOAc (250 mL) are added to the separatory funnel. After mixing, the phases are separated, and the aqueous phase is extracted with EtOAc (2 x 200 mL) (Figure 1B). The combined organic phases are sequentially washed with water (2 x 300 mL) and a saturated aqueous NaCl solution (300 mL). The organic layer is dried with MgSO4 (40 g) (Note 9), filtered through a fluted filter paper into a 1 L round-bottomed flask using a glass funnel (Note 10), and concentrated on a rotary evaporator (40 °C, 180 to 15 mmHg) (Note 11). The product is purified by crystallization by dissolving the solid in a minimal amount of EtOAc (100 mL) (Notes 12 and 13) on a gently boiling water bath. To the boiling hot solution is added, in portions, n-hexane (80 mL) (Note 14) to the point where the white precipitate that forms upon addition slowly disappears. The flask is then covered with a plastic cap, sealed with Parafilm, and the solution is allowed to cool to 24 °C on the bench and then at 4 °C in a refrigerator for 18 h (Figure 1C). The obtained solid is vacuum filtered through a round filter paper (ø = 9 cm) using a Büchner funnel and washed with a minimum amount of cold (4 °C) EtOAc/n-hexane 1:1 (approx. 20 mL) and then cold (4 °C) n-hexane (2 x 20 mL) (Note 15). The mother liquor is concentrated on a rotary evaporator (40 °C, 280 to 15 mmHg) and the remaining residue is crystallized as described above using a smaller solvent volume to obtain a second crop of material. The combined solids are dried under high vacuum (24 °C, 2 x 10-2 mm Hg) at 24 °C for 18 h to give 13.0 g (35.5 mmol, 70%) of 2 as an off-white solid (Notes 16, 17, and 18) (Figure 1D).

Figure 1. (A) Reaction mixture after addition of benzyl bromide; (B) Phases (EtOAc, top; aqueous, bottom) after shaking and allowing to separate; (C) Crystallization of 2 from a mixture of EtOAc and n-hexane; (D) Final product 2 dried under high vacuum (photos provided by submitters)
B. 1-Benzyl-7-methylene-1,5,6,7-tetrahydro-4H-benzo[d]imidazol-4-one (3). A 500 mL beaker (ø = 7 cm, h = 9 cm) (Note 19) equipped with a triangular prism-shaped stir bar (50 x 12 mm) is charged with tetra-n-butylammonium bromide (52.82 g, 163.84 mmol, 5.0 equiv) (Note 20) and tetra-n-butylammonium acetate (24.70 g, 81.92 mmol, 2.5 equiv) (Note 21). The flask is submerged in an oil bath preheated to 130 °C (Figure 2A). The salts are magnetically stirred (270 rpm) until a pale-yellow, homogenous melt is obtained (approximately 30 min) (Figure 2B) (Note 22). Then Pd(OAc)2 (220.7 mg, 0.98 mmol, 0.03 equiv) (Note 23) is added in one portion to the melt (Note 24). The mixture is stirred for 1 min (Note 25), whereupon 1-(1-benzyl-5-iodo-1H-imidazol-4-yl)pent-4-en-1-one (2) (12.00 g, 32.77 mmol, 1.0 equiv) is added slowly but continuously in one portion to the melt (Notes 24 and 26).
The resulting dark brown melt (Figure 2C) is magnetically stirred (270 rpm) for 5-10 min, at which time TLC monitoring shows complete consumption of the starting material (Note 27). The reaction is stopped by pouring the hot melt into a 1 L Erlenmeyer flask that contains a mixture of EtOAc and water (300 mL each) and is equipped with a 5 cm long stir bar that is magnetically stirred (500 rpm) at 24 °C (Note 28) (Figure 2D). Complete transfer is assured by repeatedly rinsing the reaction vessel with EtOAc and H2O from a squirt bottle. The phases are vigorously stirred for 5 min and then filtered through a firmly packed pad (2 cm) of Celite (Note 29) in a sintered glass Büchner funnel (ø = 10 cm, h = 15 cm; Porosity 4). (Figure 2E).

Figure 2: (A) The mixture of (n-Bu)4NOAc and (n-Bu)4NBr immediately after being immersed in the oil bath; (B) Appearance of the two salts after they have completely melted; (C) Appearance of the melt after adding Pd(OAc)2 and 2; (D) Hot melt being poured into a stirring mixture of EtOAc and H2O at 24 °C; (E) Filtration of the two phases through a pad of Celite using a sintered glass funnel (photos provided by submitters)
The Celite pad is washed with additional EtOAc (400 mL), and the phases are transferred into a 1 L separatory funnel and separated. The organic phase is washed with H2O (6 x 200 mL) (Figure 3A) until TLC monitoring showed no remaining tetrabutylammonium salts in the organic layer (Notes 30, 31 and 32). The organic phase is then washed with 1 M aqueous NaOH (10 x 50 mL) (Figure 3B and 3C) (Note 33 and 34) followed by a final wash with saturated aqueous NaCl solution (200 mL). The organic phase is transferred into a 1 L round-bottomed flask equipped with a 3.2 cm long oval stir bar and capped with a rubber septum. To this red solution is added activated carbon (1.0 g) (Note 35). The mixture is stirred (500 rpm) at 24 °C for 18 h and afterwards filtered through a firmly packed (2 cm) pad of Celite (Figure 3D) (Note 36) in a sintered glass Büchner funnel (ø = 10 cm, h = 15 cm; Porosity 4). This Celite pad is washed with EtOAc (400 mL) and the obtained pale, yellow-colored solution is concentrated to roughly half its volume on a rotary evaporator (40 °C, 120 mmHg). To this solution is added n-hexane (400 mL), whereupon the product precipitates (Figure 3E) (Note 37). The resultant suspension is concentrated on a rotary evaporator (40 °C, 225 to 15 mmHg) to dryness (Note 38). The yellow solid is transferred into a sintered glass Büchner funnel (ø = 6 cm, h = 6 cm; Porosity 4) (Figure 3F) (Note 39), thoroughly washed with n-hexane (500 mL) at 24 °C (Note 40), and dried under high vacuum (24 °C, 2 x 10-2 mmHg) for 18 h. Imidazole 3 (4.3 g, 18.1 mmol, 55%) is obtained as a tan-colored solid (Figure 4) (Notes 18, 41 and 42). An aromatic by-product is also produced in the reaction (Note 43).

Figure 3. (A) Washing of the organic phase with water; (B) Subsequent washing of the organic phase with 1 M NaOH; (C) Appearance of the aqueous phases (1 - 10) from basic washing procedure; (D) Filtration of the organic phase through a pad of Celite after charcoal treatment; (E) Suspension formed after half concentration of the organic phase and addition of n-hexane; (F) Washing of 3 with n-hexane using a sintered glass frit (photos provided by submitters)
Figure 4. Appearance of pure product 3 (photo provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
dimethylformamide,
potassium carbonate,
benzyl bromide,
ethyl acetate,
magnesium sulfate,
sodium chloride,
hydrochloric acid,
n-hexane,
palladium(II) acetate,
tetra-n-butylammonium bromide,
tetra-n-butylammonium acetate,
sodium hydroxide,
benzene,
methanol, and silica gel.
2. This compound was synthesized as previously described in
Org. Synth.
2021,
98, 171-193.
3.
Dimethylformamide was obtained from Sigma Aldrich (≥99.8%) and was used as received.
4.
Potassium carbonate (anhydrous, ≥99%) was obtained from Sigma Aldrich and was used as received.
5.
Benzyl bromide (98.0%, reagent grade) was obtained from Sigma-Aldrich and was used as received.
6. For reaction monitoring thin-layer chromatography was performed using silica gel F
254-plates from Merck, Darmstadt, Germany and a 1:1 mixture of
EtOAc/
n-hexane as eluent. A small sample (~50 ⊠L) was taken from the reaction mixture with a glass pipette equipped with a silicone bulb and quenched into
H2O (~1 mL) and
EtOAc (~0.2 mL) in an Eppendorf tube. The two layers were agitated by pulling multiple times into the pipette and then were allowed to separate in the tube. The organic layer was used for TLC spotting. The starting material
1 shows an R
f value of 0.2, whereas the desired product
2 shows an R
f value of 0.7 (both visible under UV light). If analysis does not indicate complete consumption of
1, another portion of
benzyl bromide (0.35 mL, 5.07 mmol, 0.1 equiv) is added, and the reaction is stirred at the same temperature for an additional 1 h - the checkers found that addition of this additional portion of
benzyl bromide was necessary.

Figure 5. TLC monitoring of completed reaction using normal phase silica plates and 1:1 EtOAc/n-hexane as eluent (left lane: starting material; right lane: reaction mixture; middle lane: co-spot) (photo provided by submitters)
7. The submitters used deionized water from their in-house supply.
8.
Ethyl acetate (≥99.8%) was obtained from Fisher Chemical and was used as received.
9.
Magnesium sulfate (97%, pure, anhydrous) was obtained from Acros Organics and was used as received.
10. The remaining
magnesium sulfate was washed with
EtOAc (25 mL).
11. The crude product obtained by benzylation of imidazole
1 mainly consists of
2 and its regioisomer
4 which are formed in a ratio of 6:1-8:1 (Figure 6).
Figure 6. Regioisomers obtained by benzylation of imidazole 2
The ratio can be determined by 1H NMR analysis of the crude product mixture after extraction, but before crystallization (diagnostic signal marked with red circle: desired product 2, 5.30 ppm; regioisomer 4, 5.50 ppm in DMSO-d6). The two regioisomers have very similar Rf values, thus the desired (major) product is best purified by crystallization.
12. A wooden stick was placed in the flask to promote boiling and prevent bumping.
13. In some cases, the volume of hot
EtOAc (100 mL) could not dissolve all of the solids. In these cases, the hot suspension was filtered through a fluted filter paper into a 500 mL round-bottomed flask using a glass funnel before continuing with the crystallization procedure. Analysis of the filtered white solid by
1H NMR showed it to be the imidazolium salt
5 (Figure 7 and see discussion).
Figure 7. Poorly soluble imidazolium salt, which results from dibenzylation
14.
n-Hexane (≥95%, HPLC grade) was obtained from Fisher Chemical and was used as received.
15. The solvent mixtures were cooled to 4 °C in a refrigerator prior to their use for washing the final compound.
16. The solid was sometimes obtained as a powder, sometimes fine needles, and sometimes thick plates (Figure 8). This variation appears to be dependent on the rate of crystallization. NMR analysis revealed that all forms are equally pure
2. The product
2 exhibits the following analytical data: R
f = 0.70 (
EtOAc/
n-hexane 1:1). mp (uncorrected): 110-111 °C.
1H NMR
pdf (600 MHz, DMSO-
d6) δ: 2.27 - 2.38 (m, 2H), 3.02 (dd,
J = 7.8, 7.0 Hz, 2H), 4.94 (d,
J = 10.4 Hz, 1H), 5.03 (d,
J = 17.1Hz, 1H), 5.30 (s, 2H), 5.85 (ddt,
J = 17.2, 10.2, 6.4 Hz, 1H), 7.14-7.15 (m, 2H), 7.30 - 7.31 (m, 1H), 7.35 - 7.37 (m, 2H), 8.23 (s, 1H).
13C NMR
pdf (150 MHz, DMSO-
d6) δ: 27.7, 37.3, 50.4, 80.6, 114.9, 127.1, 127.8, 128.7, 136.2, 137.9, 141.3, 141.4, 194.5. LC-MS (M+H) calcd for C
15H
16N
2OI (
m/z): 367. HRMS-ESI (
m/z) (provided by submitters): [2M + Na]
+ calcd for C
30H
30I
2N
4NaO
2: 755.0350, Found: 755.0357.

Figure 8. Appearance of 2 (left: needles; right: plates) from different crystallizations (photos provided by submitters)
17. Purity was determined to be 98.75% by qNMR
pdf spectroscopy in DMSO-
d6 using 36 mg of compound (
2) and 11.5 mg of mesitylene (96%) as an internal standard.
18. The product showed no decomposition after storage at 24 °C on the benchtop for one month.
19. The submitters suggest the use of a narrow beaker so that the melt has reasonable depth and so that stirring of the melt results in a vortex that facilitates the uptake and mixing of the added materials.
20.
Tetra-n-butylammonium bromide (≥99.0%, reagent plus) was obtained from Sigma-Aldrich and was used as received.
21.
Tetra-n-butylammonium acetate (≥90%, technical) was obtained from TCI Chemicals and was used as received.
22. The remaining salt residues sticking to the wall of the beaker were brought into the melt by carefully heating with a heat gun.
23.
Palladium(II) acetate (Pd(OAc)2 (recrystallized, 97%)) was obtained from Sigma-Aldrich and was used as received.
24. Care should be taken to carefully pour the material into the middle of the vortex to ensure that it is readily taken up by the melt and does not remain floating upon the surface.
25. The melt became pale brown with a few remaining undissolved particles of
Pd(OAc)2.
26. In the event that
2 was obtained as thick plates, the solid was finely ground using a mortar and pestle before addition.
27. Completion of the reaction occurred rapidly after dissolution of the starting material
2 into the melt. The submitters recommend to monitor the reaction by performing TLC-analyses every two minutes after the addition of
2. A sample is taken by putting the tip of a glass pipette into the hot reaction mixture and the black residue remaining on the glass pipette is diluted with
EtOAc and then used for TLC monitoring. Thin-layer chromatography was performed using silica gel 60 F
254-plates from Merck, Darmstadt, Germany and a 9:1 mixture of CH
2Cl
2/
MeOH as eluent. The starting material
2 shows an R
f value of 0.91, whereas the desired product
3 shows an R
f value of 0.46 (both visible under UV light) (Figure 9).

Figure 9. TLC monitoring of the Heck reaction immediately after adding 2 (left plate) and 5 min after the addition (right plate). Left lanes: starting material 2; Right lanes: reaction mixture diluted with EtOAc; middle lanes: co-spot. CH2Cl2/MeOH (9:1) used as eluent (photos provided by submitters)
28. The submitters wrapped paper towels around the beaker to facilitate pouring of the hot melt. This also assures that no oil sticking to the outside of the beaker is unintentionally transferred into the reaction mixture.
29. Celite Standard Super Cel was obtained from Carl Roth and was used as received.
30. The tetrabutylammonium salts don't exclusively partition into the aqueous layer when using
EtOAc as an organic solvent, which necessitates multiple washes. The submitters suggest monitoring the organic layer for the presence of remaining salts by thin-layer chromatography (TLC) (CH
2Cl
2/
MeOH 9:1). The salts appear as a streaky spot (visible with I
2 stain, but not UV) below the product spot (visible under UV and with I
2 stain). Washing should be repeated (usually 5-7 times) until no further tetrabutylammonium salts can be detected (Figure 10).
Figure 10. TLC monitoring of the washing procedure with H2O. From left to right lane: organic layer after the first phase separation, then after the next six aqueous washes; CH2Cl2/MeOH 9:1 used as eluent; TLC plate is stained with iodine (photos provided by submitters)
31. Accurate phase separation is occasionally difficult due to cloudiness and bubble formation between the layers. The submitters prefer to keep a minimal amount of water in the funnel during each separation so as not to lose product during the washing procedure.
32. After the washing is complete, a small volume of the organic phase is removed and concentrated on a rotary evaporator (40 °C, 6 mmHg). The residue is analyzed by
1H NMR spectroscopy in order to determine the ratio of desired product
3 and its aromatized isomer
6 (Figure 11 and see Discussion). This ratio varies between 5:1-8:1 (diagnostic signal marked with red circle: desired product
3, 5.30 ppm; aromatized byproduct
6, 5.53 ppm in CDCl
3).
Figure 11. Products arising from Heck reaction of iodoimidazole 2
33.
Sodium hydroxide was obtained from VWR Chemicals Prolabo and was used as received.
34. These washes remove hydroxybenzimidazole
6 from the organic layer by deprotonating the hydroxyl group and helping to partially partition
6 into the aqueous layer. Ketone
3 also partially partitions into the aqueous layer during this procedure, but material loss is minimal. That both products partition into the aqueous phase is seen visually as depicted in Figure 3C: Upon standing for a few minutes at 24 °C, a fine off-white solid (
3 and
6) precipitates from the aqueous phase.
The washing procedure can be monitored by thin-layer chromatography using silica gel 60 F254-plates from Merck, Darmstadt, Germany and benzene/MeOH (9:1) as eluent (Figure 12). Byproduct 6 appears as a slightly faster eluting spot than 3 under these conditions. Small quantities of byproduct 6 visualize very well under UV and with I2 staining, making it difficult to determine when to stop washing. The submitters have found that 10 washes consistently provide sufficiently pure samples of 3 (also see discussion), and recommend using this number of washes, but still monitoring via TLC for a visual analysis. After the 10 washes, a small volume from the organic phase can be concentrated and analyzed by 1H NMR. If a 3/6 ratio ≥50:1 is not obtained after 10 washes, further washing with 1M NaOH must be performed.

Figure 12. TLC monitoring of the basic washing procedure with 1M NaOH. From left to right lane: pure product 3 obtained in a different experiment, organic phase from 1st to 5th wash (left TLC plate) and organic phase from 6th to 10th wash (right TLC plate), benzene/MeOH 9:1 used as eluent; TLC plate is stained with iodine (photos provided by submitters)
35. Activated carbon was obtained from Sigma-Aldrich and was used as received.
36. Treatment of the organic phase with charcoal was done to decolorize the product by removing trace impurities such as palladium.
37. The submitters adopted this precipitation procedure due to difficulties in removing all traces of
EtOAc from
3 when it was concentrated to a solid from an
EtOAc solution, even after the resulting solid was dried at 100 °C under high vacuum for 24 h. This precipitation method reliably provided solvent-free
3 after drying the filtered solid under high vacuum.
38. If the product re-dissolves during concentration, the flask should be removed from the rotary evaporator and additional
n-hexane should be added, to ensure that the product remains a solid throughout this process.
39. The majority of the solid can be scratched out of the flask with a spatula and transferred to the funnel by rinsing with
n-hexane. Any solid remaining in the round-bottomed flask is negligible, but if a quantitative transfer is desired, it can be redissolved in
EtOAc, transferred into a smaller (100 mL) round-bottomed flask, concentrated on a rotary evaporator (40 °C, 150 to 35 mmHg) to a solid, and then transferred to the Büchner funnel.
40. To ensure complete removal of trace
EtOAc, the solid was crushed in the funnel with a spatula, then covered with
n-hexane, the resulting suspension was well mixed with the spatula, and then vacuum was applied to pull the solvent through the funnel. This procedure was repeated (3-4 times) until 500 mL of
n-hexane were consumed.
41. The product (
3) exhibits the following analytical data: R
f = 0.46 (CH
2Cl
2/
MeOH 9:1) and 0.19 (
benzene/
MeOH 9:1). mp (uncorrected): 147.0-149.0 °C.
1H NMR
pdf (600 MHz, CDCl
3) δ: 2.70 (t,
J = 6.6 Hz, 2H), 2.81 (t,
J = 6.0 Hz, 2H), 5.13 (s, 1H), 5.26 (s, 1H), 5.33 (s, 2H), 7.08 (d,
J = 7.2 Hz, 2H), 7.35 - 7.40 (m, 3H), 7.57 (s, 1H).
13C NMR
pdf (151 MHz, CDCl
3) δ: 34.1, 38.6, 49.7, 112.2, 126.3, 128.4, 129.3, 132.9, 134.9, 138.0, 139.4, 141.9, 191.5. Purity was determined to be 89.6% by qNMR
pdf spectroscopy in CDCl
3 using 23.9 mg of compound (
3) and 10.7 mg of dimethyl sulfone (99.65%) as an internal standard. LC-MS (M+H) calcd for C
15H
15N
2O (
m/z): 239. HRMS-ESI (
m/z) (provided by submitters): [2M + Na]
+ calcd for C
30H
28N
4NaO
2+: 499.2104, found: 499.2109.
42. While
3 obtained via this extractive purification is 89.6% pure (based on qNMR), a sample that is entirely free of
6 and other minor impurities can be prepared by crystallization: a sample of
3 (1.0 g, 4.19 mmol) is dissolved in hot
EtOAc (~40 mL) on a water bath (100 °C), filtered while hot, and
n-hexane (~15 mL) is then added while the solution is still on the water bath until the white precipitate which forms only slowly disappears. The mixture is cooled to 24 °C and then to -20 °C for 18 h. After filtration through a round paper filter (ø = 5.5 cm) using a Büchner funnel, washing with cold (4 °C)
EtOAc/
n-hexane 1:1 (13 mL) and then cold (4 °C)
n-hexane (2 x 15 mL), and air drying for 1 h on the filter using house vacuum, an off-white solid (0.774 g, 3.23 mmol, 77.4%) was obtained (Figure 13).
1H NMR
pdf (600 MHz, CDCl
3) δ: 2.70 (t,
J = 6.6 Hz, 2H), 2.81 (t,
J = 6.0 Hz, 2H), 5.13 (s, 1H), 5.26 (s, 1H), 5.34 (s, 2H), 7.08 (d,
J = 7.2 Hz 2H), 7.33 - 7.40 (m, 3H), 7.57 (s, 1H).
13C NMR
pdf (151 MHz, CDCl
3) δ: 34.1, 38.6, 49.7, 112.2, 126.3, 128.4, 129.3, 132.9, 134.9, 138.0, 139.4, 141.9, 191.5. Purity was determined to be 96.8% by qNMR
pdf spectroscopy in CDCl
3 using 24 mg of compound (
3) and 9.7 mg of dimethyl sulfone (99.65%) as an internal standard.

Figure 13. Pure 3 obtained by crystallization from hot EtOAc/n-hexane
43. The submitters obtained an analytical sample of aromatized byproduct
6 as follows: The aqueous phases of the basic washing procedure (see
Note 34) are combined and acidified by dropwise addition of concentrated
HCl to pH~5-6. Then the aqueous phase is extracted with
EtOAc (3 x 200 mL), whereupon the combined organic phases are washed with brine (300 mL) and concentrated on a rotary evaporator (40 °C, 280 to 15 mmHg). The obtained brown solid is suspended in hot
EtOAc (50 mL). The suspension is filtered using a fluted filter paper in a 250 mL round-bottomed flask, and to the hot solution of
EtOAc is added
n-hexane (50 mL). The flask is covered with a plastic cap (wrapped with Parafilm) and allowed to cool to 24 °C on the bench and then at -20°C in a freezer. The resulting yellow solid is filtered through a round filter paper (ø = 4.5 cm) using a Büchner funnel and washed with a minimum amount of cold (4 °C)
EtOAc/
n-hexane 1:1 (20 mL) and then cold (4 °C)
n-hexane (2 x 20 mL) and air dried on the filter for 1 h using house vacuum (Figure 14).

Figure 14. Appearance of aromatized byproduct 6 (photo provided by submitters)
The aromatized byproduct 6 exhibits the following analytical data (provided by the submitters): Rf = 0.46 (CH2Cl2/MeOH 9:1) and 0.30 (benzene/MeOH 9:1). mp (uncorrected) 207-209 °C (decomposition). 1H NMR (400 MHz, DMSO-d6) δ 2.26 (s, 3H), 5.64 (s, 2H), 6.44 (d, J = 7.8 Hz, 1H), 6.67 (dd, J = 7.9, 1.0 Hz, 1H), 6.92 - 6.98 (m, 2H), 7.20 - 7.37 (m, 3H), 8.15 (s, 1H), 9.57 (br. s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 17.4, 48.8, 106.5, 111.7, 125.2, 125.6, 127.5, 128.9, 133.9, 134.1, 139.2, 144.0, 147.6. HRMS-ESI (m/z): [2M + Na]+ calcd for C30H28N4NaO2+: 499.2104, Found: 499.2110.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
We required an efficient synthesis of ketone
3 and related substances for a project devoted to the synthesis of covalent methionine aminopeptidase 2 (MetAP2) inhibitors. Our first-generation library of substances were spiroepoxyimidazoles
7, which were all synthesized from
1 via alkylation, Heck cyclization, and epoxidation with DMDO (Figure 15A). The compounds were designed to mimic the natural product ovalicin, a potent and selective covalent MetAP2 inhibitor (Figure 15B). Our second-generation inhibitors were based on a triazole scaffold, and we found them to be more potent and to have better physicochemical properties than the imidzoles (Figure 15C).
2Figure 15. (A) Synthesis route to 1st generation MetAP2 inhibitors; (B) Small library of putative MetAP2 inhibitors inspired by ovalicin; (C) General structure of 2nd generation MetAP2 inhibitors
Outside of the context of our MetAP2 inhibitor project, compound
3 is an interesting building block due to the fact that it can potentially be derivatized in a variety of ways to generate a diverse collection of substances (Scheme 1). Besides oxidation to epoxide
7 as mentioned above, ketone
3 can also be transformed to aziridines
8,
3 undergo reductive amination to give amines
9,
4 or reduced to alcohol
10.
5 Alternatively, the exocyclic double bond can be reduced to a methyl group to afford ketone
11,6 or undergo Huisgen cycloaddition reactions with 1,3-dipoles to afford spirocycles of type
12.
7 Many more manipulations can also be envisaged, including those that involve removal of the benzyl group and replacement with other substituents.
8Scheme 1. Some possible transformations with annulated imidazole 3
We now discuss some noteworthy details from our explorations into the synthesis of 3. In the first step, iodoimidazole 1 is alkylated with benzyl bromide. Because the two imidazole nitrogens in 1 are regiotopic, two products (2 and 4) can be expected and, indeed, are formed in the mono-benzylation reaction (Scheme 2).
Scheme 2. Reaction of 1 with benzyl bromide produces two regioisomeric products
We investigated the influence of temperature, solvent, base, and electrophile on the reaction and found
benzyl bromide with K
2CO
3 in DMF to give a clean, rapid conversion to the two products. Conducting the reaction at 0 °C gave the most consistent product ratios, with
2 and
4 formed in a ratio between 6:1 and 8:1. Because
2 is purified by crystallization, the resulting mother liquor is highly enriched in
4. We speculated that if the product ratio obtained in the initial reaction reflected a thermodynamic ratio, we could potentially equilibrate the mother liquor mixture, via the intermediacy of the corresponding dibenzylated imidazolium salt (
5), by heating with a catalytic amount of
benzyl bromide (Scheme 3).
9 Attempts to perform such an equilibration revealed that
5 forms readily and is quite stable; therefore, no equilibration occurred. Moreover, running the initial benzylation reaction with larger equivalents of
benzyl bromide (
e.g. 1.2 equiv), even at lower temperatures, results in significant imidazolium salt formation. Compound
5 is highly crystalline and can cause problems during the crystallization of
2 (see Notes
11 and
13). Therefore, the submitters advise using only a slight excess of
benzyl bromide (1.01-1.05 equivalents) for the alkylation of imidazole
1.
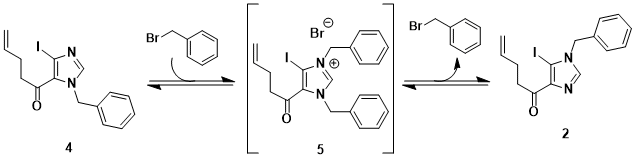
Scheme 3. Envisioned equilibration of 2 and 4 via imidazolium salt 5
The subsequent intramolecular Heck cyclization with
2 proved challenging. Our initial attempts with "typical" Heck conditions, showed that only ligandless Jeffery conditions
(Pd(OAc)2, (
n-Bu)
4NBr, Na
2CO
3, MeCN/
H2O, 70 °C) resulted in any reaction. However, long reaction times (several days) and multiple catalyst loadings were required to obtain useful quantities of product, and we never observed complete consumption of the starting material. We were, therefore, extremely pleased to find that the method of Nacci and co-workers,
11 which employs palladium colloids/nanoparticles in an ionic liquid of (
n-Bu)
4NBr and (
n-Bu)
4NOAc at 130 °C worked extremely well, with full conversion of the starting material in less than 10 minutes.
In addition to the desired product 3, we observed its aromatized tautomer hydroxybenzimidazole 6 as a byproduct, typically with a 3/6 ratio between 5:1 and 8:1 (Scheme 4). We were pleased that 3 was stable enough to be isolated, but were concerned that isomerization of 3 to 6 was taking place during the reaction. If this was the case, small changes in the time spent in the reaction melt at 130 °C could produce variable product ratios. To investigate our concerns, we treated pure 3 under both strongly basic and acidic conditions, and found essentially no isomerization. We also found that performing the Heck reaction for longer reaction times, or under an inert anhydrous atmosphere, or with an added 10 equivalents of water had no effect on the 3/6 product ratio. We also re-subjected pure 3 to the reaction conditions for 1 h, but no 6 was detected by NMR of the reaction mixture upon work-up.
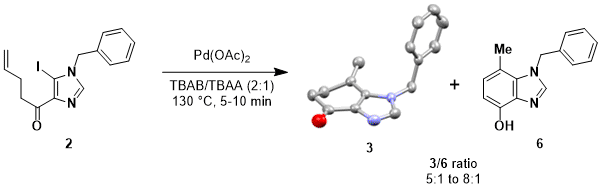
Scheme 4. Heck reaction of iodoimidazole 2 affords the desired product (3) (X-ray structure)12 and its aromatized congener (6). Ellipsoids for 3 are shown at a 50% probability level; carbon: gray, nitrogen: blue, oxygen: red
Since the
3/
6 ratio is unaffected by the presence of air or water, and
3 does not isomerize under the reaction conditions, we postulate that
6 is formed via a side reaction involving the intermediate palladium hydride species formed during the Heck reaction. The catalytic cycle starts with the oxidative addition of Pd(0)
(Pd(OAc)2 can be reduced to Pd(0) under these conditions to form colloids/nanoparticles, which are productive Heck catalysts)
13 into the I--Ar bond of
2 to form
13 (Scheme 5). After coordination, Pd(II) inserts into the double bond, giving intermediate
14.
⊠-Hydride elimination forms the expected product
3 and H-Pd-I. Reductive elimination of the latter regenerates the catalyst Pd(0), and produces HI, which is sequestered by acetate.
Scheme 5. Catalytic cycle of Heck reaction with iodoimidazole 2 affording product 3 (including postulated reaction path to hydroxybenzimidazole 6)
If H-Pd-I re-inserts into
3 before undergoing reductive elimination, intermediate
15 could be formed.
14 A subsequent "productive"
syn-
β-hydride elimination from
15 would yield
16, and after keto-enol tautomerization, hydroxybenzimidazole
6. Compound
6, due to its extended aromaticity, is assuredly more thermodynamically stable than
3 and will not revert to
3. This mechanistic explanation for the conversion of
3 into
6 can only take place in the presence of a palladium hydride species, and is consistent with our observations,
i.e. after consumption of the starting material (or if no starting material is present to begin with), palladium hydride species should not be formed, and isomerization of
3 to
6 cannot take place.
Isomers 3 and 6 are almost co-polar by TLC under all solvent systems that we tested, making chromatography ineffective as a means of purification. Crystallization was also problematic, due to the relatively low solubility and high crystallinity of 6. We, therefore, employed a basic extraction procedure, which enabled enrichment of 3 in the organic phase due to the relatively low pKa of hydroxybenzimidazole 6. In one full-scale run of this reaction, we quantified the success of the extraction procedure by taking aliquots of the organic phase after the first, third, fifth, seventh and tenth washing step (see Note 34). These were concentrated on a rotary evaporator and measured by NMR to evaluate the 3/6 ratio at each step of this procedure. As can be seen in Figure 16, 10 washes were sufficient to bring the content of 6 to ~2%. Because each basic wash also leads to a loss of desired product 3 (monitored by TLC of the aqueous phase), the washing procedure should be stopped after ten cycles.
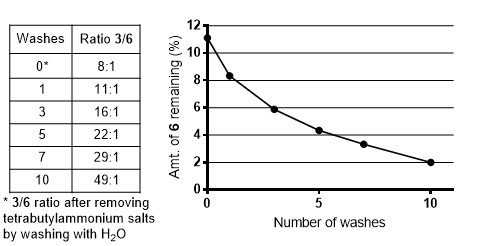
Figure 16. Product ratio 3/6 in the organic phase throughout the basic wash as determined by NMR in table and graph formats
Appendix
Chemical Abstracts Nomenclature (Registry Number)
1-(5-Iodo-1H-imidazol-4-yl)pent-4-en-1-one: 4-Penten-1-one, 1-(5-iodo-1H-imidazol-4-yl)-; (1) (1585257-59-5)
Potassium carbonate: Carbonic acid, potassium salt (1:2); (584-08-7)
Benzyl bromide: Benzene, (bromomethyl)-; (100-39-0)
1-(1-Benzyl-5-iodo-1H-imidazol-4-yl)pent-4-en-1-one: 4-penten-1-one, 1-[5-iodo-1-(phenylmethyl)-1H-imidazol-4-yl]-; (2) (1585257-17-5)
Palladium(II) acetate: Acetic acid, palladium(2+) salt (2:1); (3375-31-3)
Tetra-n-butylammonium bromide: 1-Butanaminium, N,N,N-tributyl-, bromide (1:1); (1643-19-2)
Tetra-n-butylammonium acetate: 1-Butanaminium, N,N,N-tributyl-, acetate (1:1); (10534-59-5)
1-Benzyl-7-methylene-1,5,6,7-tetrahydro-4H-benzo[d]imidazole-4-one: 4H-Benzimidazol-4-one, 1,5,6,7-tetrahydro-7-methylene-1-(phenylmethyl)-; (3) (1585257-23-3)

|
Michael Morgen received his Ph.D. in inorganic chemistry from the Ruprecht-Karls-University, Heidelberg in 2013 while working with Prof. Dr. Peter Comba on the design of novel bispidine-based multimodal imaging agents. Afterwards he joined the Cancer Drug Development Group of Dr. Aubry Miller and Dr. Nikolas Gunkel at the German Cancer Research Center (DKFZ). His current research interests lie in the design and synthesis of organic and metal-organic inhibitors for cancer-related drug targets. |

|
Jasmin Lohbeck was trained as chemistry laboratory assistant at BASF in Ludwigshafen, Germany, from 2006 - 2009. She subsequently joined the chemistry lab of Dr. Aubry Miller at the German Cancer Research Center and specialized in preparative organic synthesis. |

|
Aubry Miller leads the Cancer Drug Development Group at the German Cancer Research Center (DKFZ) in Heidelberg, Germany. His laboratory focuses on the discovery and biological characterization of chemical probes for use as tools and drug leads in cancer research. |

|
Nageswara Rao Kalikinidi earned his Ph.D. in 2018 in Synthetic Organic Chemistry from the CSIR-IICT, India, under the guidance of Dr. Subhash Ghosh. His research focused on developing a synthetic route for the total synthesis of biologically active and architecturally complex marine natural products. In 2019, he joined in Prof. E. J. Corey research group at Harvard University, where his research was focused on the enantioselective epoxidation reactions with chiral azatetracycle catalysts. After completing his postdoctoral studies in 2021, Nageswara joined TCG GreenChem, Inc. as a post-doctoral Scientist. His research interests include the design and development of green and robust large-scale production of active pharmaceutical ingredients (APIs) and synthetic methodology of novel organic transformations using chiral phosphine ligands. |

|
Venumadhav Janganati earned his Ph.D. in synthetic organic chemistry from National Institute of Technology, Warangal, India in 2009 under the guidance of Prof. B. Rajitha. His research focused on synthesis of bioactive heterocyclic molecules and developing novel methodologies by using new reagents. In his postdoctoral training at the University of Arkansas for Medical Sciences, USA, he designed and synthesized natural product (parthenolide) derivatives for anti-cancer therapy, and he was also involved in design and synthesis of lobelane derivatives, which focused on the development of new treatments for methamphetamine addiction. Currently working as a senior scientist in Process Chemistry at TCG GreenChem, Inc., his research interests include the process research and development and asymmetric catalysis. |

|
Dr. Gopal Sirasani received his Bachelor's and Master's degrees in Hyderabad, India. He obtained his Ph.D. in synthetic organic chemistry in 2011 from Temple University, Philadelphia under the guidance of Prof. Rodrigo B. Andrade. His doctoral research focused on developing novel methodologies, total syntheses of natural products and their analogs thereof. He got his post-doctoral training in the laboratory of Prof. Emily Balskus at Harvard University, where he developed biocompatible organic reactions utilizing microbially generated reagents to realize transition metal catalysis in the presence of microbes. In 2013, Gopal began his industrial career at Melinta Therapeutics, New Haven, CT. He is currently working at TCG GreenChem, Inc. as a Director in the department of process research and development. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved