Checked by Bogdan R. Brutiu, Martina Drescher, Daniel Kaiser and Nuno Maulide
1. Procedure (Note 1)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available
via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
(2S)-bornane-2,10-sultam,
tetrahydrofuran, 1.6 M
n-Butyllithium,
hexanes,
bromoacetyl bromide,
diethyl ether,
sodium sulfate,
N,N-dimethylformamide,
sodium azide,
methanol, conc. HCI, 10 wt.%
palladium on carbon,
sodium bicarbonate and
dichloromethane as well as the proper procedures for handling, addition, cannulation and quenching of highly reactive
n-butyllithium and
bromoacetyl bromide, handling and addition of potentially explosive
sodium azide and hydrogenolysis reaction using
palladium on carbon.
2. A 100 mL dropping funnel would be ideal for the experiment since it reduces the measuring error caused by filling
n-butyllithium two times to make up the required volume.
3. The reaction is very sensitive to moisture and air. Hence, it was carried out with extreme care, under an inert (argon) environment. All the joints in the experimental setup were sealed using parafilm tape.
4.
(2S)-Bornane-2,10-sultam (98%) was purchased from AK scientific and used as received. The checkers purchased this compound from Fluorochem.
5.
Tetrahydrofuran (certified ACS grade) was purchased from Fisher Scientific and was distilled from Na/benzophenone under argon. The reaction is very sensitive to water, therefore care was taken to use dry solvents. The checkers purchased anhydrous
THF from TCI.
6. 1.6 M
n-butyllithium in
hexanes was purchased from Sigma-Aldrich and used as received.
7. Weight of the 1.6 M
n-butyllithium used is given based on the
n-butyllithium moles.
8. After complete addition of
n-butyllithium in
hexanes, the pressure-equalizing dropping funnel can be quickly replaced with a rubber septum at an increased argon flow.
9.
Bromoacetyl bromide (≥98%) was purchased from Sigma-Aldrich and used as received.
10. The reaction was monitored by TLC using silica gel HL TLC plates, purchased from Sorbent Technologies, Inc. The plate was developed using (3:1, v/v)
hexanes-
ethyl acetate as the mobile phase and visualized using cerium ammonium molybdate stain (Figure 8). The starting material forms a deep-blue colored spot and the product forms a gray-blue colored spot on the TLC upon heating with the stain. The R
f value of the starting material
1 was 0.26 and the R
f of the product
2 was 0.41.
Figure 8. TLC analysis of the reaction mixture at 2 h, after addition of bromoacetyl bromide (photo provided by submitter)
11. Addition of water was done slowly and while the reaction mixture was chilled. Even though water can react vigorously with the small amount of excess
bromoacetyl bromide and/or
n-butyllithium remaining in the reaction mixture, such was not observed during the dilution.
12.
Diethyl ether, anhydrous (BHT stabilized, certified ACS grade) was purchased from Fisher Scientific and used as received.
13.
Sodium sulphate, anhydrous was purchased from MilliporeSigma and used as received.
14. The crude product is of sufficient purity for use in Step B, therefore purification is not required at this stage. The presence of a small amount of bromoacetic acid was occasionally observed in the product by
1H NMR and/or a crude weight is occasionally determined to be greater than the theoretical value. If this is the case, partitioning the crude material between
NaHCO3 (sat. aq.) (100 mL) and
diethyl ether (500 mL) can address those issues. In fact, this route to synthesize Oppolzer's
glycylsultam does not require any chromatographic or crystallization purification apart from a simple organic wash after the hydrogenolysis at the final step. Hence crude
2 was taken directly to the next step.
15. Characterization data for the crude product
2 (data provided by checkers):
1H NMR
pdf (400 MHz, CDCl
3) δ: 4.32 (d,
J = 13.1 Hz, 1H), 4.19 (d,
J = 13.1 Hz, 1H), 3.90 (dd,
J = 7.6, 5.1 Hz, 1H), 3.49 (app q,
J = 13.8 Hz, 2H), 2.19-2. 02 (m, 2H), 2.00-1.82 (m, 3H), 1.53-1.31 (m, 2H), 1.14 (s, 3H), 0.97 (s, 3H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 164.6, 65.6, 52.8, 49.1, 48.0, 44.6, 38.0, 32.9, 27.6, 26.5, 20.8, 20.0; IR (neat): 2989, 2959, 2884, 2360, 2341, 2256, 1696, 1616, 1541, 1507, 1481, 1456, 1435, 1411, 1395, 1373, 1327, 1313, 1301, 1258, 1232, 1205, 1167, 1152, 1132, 1110, 1084, 1059, 1039, 987, 952, 939, 911, 888, 868, 843, 805, 774 cm
-1; HRMS (ESI) calcd for C
12H
18NO
3SBrNa [M + Na]: 358.0083. Found: 358.0092. Purity of the crude product (
2) was assessed to be 78% based on qNMR
pdf analysis using
1,3,5-trimethoxybenzene as an internal standard. The checkers performed a second run on half scale, yielding 23.7 g (89% yield, 88% purity). The submitters reported the following result on a full scale reaction: 47.7 g, 82% yield, 81% purity.
16. The major impurity present in crude
2 is the unreacted camphor sultam
1 (Figure 8), which is completely inert towards ancillary transformations. But if further purification is required, flash chromatography can be performed to obtain pure
2 as follows: Silica gel 60 Å was purchased from Sorbent Technologies, Inc. The flash column (dimensions: 13 mm inner diameter, 203 mm in height) is wet packed with silica gel (15 g; 150 mm, height of the silica bed in the column) using a solution of (4:1, v/v)
hexanes-
ethyl acetate. Crude
bromoacetylsultam 2 (59 mg, dissolved in 1 mL of methylene chloride) is wet loaded onto the packed silica column. The column is eluted with 100 mL of (4:1, v/v)
hexanes-
ethyl acetate and then continued with 200 mL of (3:1, v/v)
hexanes-
ethyl acetate until the product completely elutes off the column (Figure 9). The fraction collection is begun (5-mL fractions) immediately after starting the solvent elution and each fraction is analyzed by TLC to find the product (Figure 9). The plate was developed using (3:1, v/v)
hexanes-
ethyl acetate as the mobile phase and visualized using cerium ammonium molybdate stain followed by heating. Column fractions 7-9 were combined and concentrated by rotary evaporation (35 ℃, started with 50 mmHg and gradually lowered to 10 mmHg) and then at 0.1 mmHg to afford purified
2 (49.5 mg).
Figure 9. TLC analysis of the column chromatography fractions of 2 (photo provided by submitter)
17. Pure
bromoacetylsultam 2 has the following characteristics (data provided by submitters): mp 105-107 ℃;
1H NMR (500 MHz, CDCl
3) δ: 4.34 (d,
J = 12.8 Hz, 1H), 4.20 (d,
J = 13.0 Hz, 1H), 3.91 (dd,
J = 7.8, 5.0 Hz, 1H), 3.53 (d,
J = 13.8 Hz, 1H), 3.46 (d,
J = 13.9 Hz, 1H), 2.18-2.12 (m, 1H), 2.09 (dd,
J = 14.0, 7.6 Hz, 1H), 1.96-1.87 (m, 3H), 1.47-1.40 (m, 1H), 1.39-1.32 (m, 1H), 1.16 (s, 3H), 0.98 (s, 3H);
13C NMR (126 MHz, CDCl
3) δ: 164.7, 65.6, 52.9, 49.2, 48.0, 44.7, 38.1, 32.9, 27.7, 26.6, 20.9, 20.0; HRMS (ESI)
m/z calcd for C
12H
19BrNO
3S [M+H]
+ 336.0269; found, 336.0282; IR (film): 3010, 2958, 2907, 2881, 1701, 1458, 1412, 1390, 1368, 1332, 1311, 1264, 1232, 1208, 1166, 1133, 1084, 1063, 1038, 988, 952, 939, 909, 889, 868, 804, 777, 745, 693, 632, 618, 563, 537, 529, 495, 456 cm
-1.
18. The number of moles of
bromoacetylsultam 2 in the crude is calculated based on the purity percentage from the quantitative NMR data.
19.
N,N-Dimethylformamide (certified ACS grade) was purchased from VWR analytical and used as received. The checkers purchased
DMF from Acros.
20.
Sodium azide (>99.0%) was purchased from TCI chemicals and used as received. The checkers purchased this compound from Sigma Aldrich.
21. Solid
sodium azide was added to the reaction using a plastic spatula. Metal spatulas can react with
sodium azide and produce explosive metal azides. Hence, care was taken to minimize the contact with metals.
22. The reaction is difficult to monitor using TLC (Figure 10). The starting material
2 and the product
3 have the same R
f value in the given solvent system. The TLC plate was developed using (3:1, v/v)
hexanes-
ethyl acetate mobile phase and visualized using ninhydrin stain. The R
f value of both
2 and
3 was 0.40. Despite having the same Rf value, the compounds stain differently when visualized using the ninhydrin stain. Starting material (
2) forms a faint pink colored spot and the product (
3) forms an orange-pink colored spot on the TLC upon heating.
Figure 10. TLC analysis of the starting material 2 and the product 3 (photo provided by submitter)
Hence, the reaction was monitored using 1H NMR. In most parts of the spectrum, the NMR chemical shifts of the starting material 2 and the product 3 are quite similar. Distinctive differences were mostly observed between the AB quartets in 4.50-3.30 ppm range, hence, these signals were used to monitor the progress of the reaction. The reaction usually goes to the completion within 5-6 h. To confirm the complete conversion, 1H NMR analysis was performed on the reaction mixture after 8 h, including NMR analysis of the reaction mixture that was spiked with the starting material (Figure 11). To prepare the NMR sample, approximately 0.3 mL of the reaction mixture was removed and partitioned between 0.5 mL of water and 0.5 mL of (1:1, v/v) diethyl ether-hexanes in a 4 mL vial. The organic phase was removed using a pasture pipette and concentrated under vacuum (10 mmHg). The NMR sample was prepared in CDCl3 and monitored using a Varian 400 MHz spectrometer.
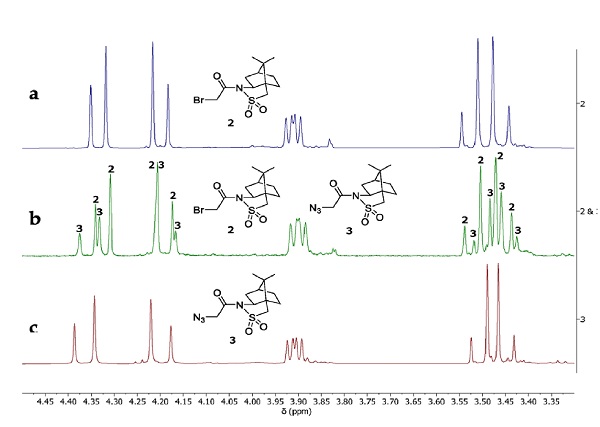
Figure 11. NMR analysis of (a) starting material 2, (b) reaction mixture after 8 h, spiked with the starting material, (c) product 3 (provided by submitter)
23. Purification was not required at this stage and crude
3 was directly taken to the next step.
24. Characterization data for the crude product
3 (data provided by checkers):
1H NMR
pdf (400 MHz, CDCl
3) δ: 4.35 (d,
J = 17.3 Hz, 1H), 4.18 (d,
J = 17.3 Hz, 1H), 3.90 (dd,
J = 7.7, 5.0 Hz, 1H), 3.50 (d,
J = 14.1 Hz, 1H), 3.45 (d,
J = 14.2 Hz, 1H), 2.32-2.14 (m, 1H), 2.10 (dd,
J = 14.0, 7.9 Hz, 1H), 2.00-1.83 (m, 3H), 1.47-1.31 (m, 2H), 0.97 (s, 3H), 1.12 (s, 3H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 166.7, 65.4, 52.8, 51.4, 49.4, 48.0, 44.7, 38.2, 32.9, 26.5, 20.8, 19.9; IR (film): 2961, 2359, 2337, 2160, 2109, 1992, 1703, 1541, 1458, 1415, 1376, 1332, 1269, 1238, 1220, 1167, 1137, 1117, 1066, 1040, 985, 941, 872, 808, 781 cm
-1; HRMS (ESI) calcd for C
12H
18N
4O
3SNa [M + Na]: 321.0992. Found: 321.1001. The purity of the crude product
3 was assessed to be 84% based on qNMR
pdf analysis using
1,3,5-trimethoxybenzene as an internal standard. The checkers performed a second run on half scale, yielding 17.7 g (92% yield, 96% purity). The submitters reported the following result: 36.3 g, 88% yield, 83% purity.
Percentage yield calculation,
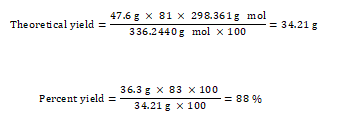
25. The major impurity that is present in crude
3 is again the unreacted camphorsultam
1 carried over from step A, which is completely inert towards ancillary transformations. But if desired, flash chromatography can be performed to obtain pure
2 as follows: The flash column (dimensions: 13 mm inner diameter, 203 mm in height) is wet packed with silica gel (15 g) using a solution of (4:1, v/v)
hexanes-
ethyl acetate (150 mm, height of the silica bed in the column).
Azidoacetylsultam 3 (52.0 mg, dissolved in 1 mL of methylene chloride) is wet loaded onto the packed silica column. The column is eluted with 100 mL of (4:1, v/v)
hexanes-
ethyl acetate and then continued with 200 mL of (3:1, v/v)
hexanes-
ethyl acetate until the product completely eluted off the column. The fraction collection is begun (5-mL fractions) immediately after starting the solvent elution and each fraction is analyzed by TLC to find the product (Figure 12). The plates were developed using (3:1, v/v)
hexanes-
ethyl acetate as the mobile phase and visualized using both ninhydrin and cerium ammonium molybdate stain followed by heating (camphorsultam
1 is insensitive to ninhydrin stain). Column fractions 7-10 were combined and concentrated by rotary evaporation (35 ℃, starting with 50 mmHg and gradually lowering to 10 mmHg) and then at 0.1 mmHg to afford purified
3 (45.0 mg).

Figure 12. TLC analysis of the column chromatography fractions of 3, with (a) ninhydrin stain and (b) cerium ammonium molybdate stain (photos provided by submitter)
26. Pure
azidoacetylsultam 3 has the following characteristics (data provided by submitters): mp 79-81 ℃;
1H NMR (500 MHz, CDCl
3) δ: 4.36 (d,
J = 17.3 Hz, 1H), 4.20 (d,
J = 17.3 Hz, 1H), 3.91 (dd,
J = 7.9, 4.9 Hz, 1H), 3.51 (d,
J = 13.9 Hz, 1H), 3.45 (d,
J = 13.8 Hz, 1H), 2.21 (dq,
J = 14.0, 3.8 Hz, 1H), 2.12 (dd,
J = 14.0, 7.9 Hz, 1H), 1.96-1.87 (m, 3H), 1.47-1.41 (m, 1H), 1.40-1.34 (m, 1H), 1.14 (s, 3H), 0.98 (s, 3H);
13C NMR (126 MHz, CDCl
3) δ: 166.7, 65.4, 52.8, 51.5, 49.4, 48.0, 44.7, 38.2, 32.9, 26.5, 20.9, 20.0 ; HRMS (ESI)
m/z calcd for C
12H
19N
4O
3S [M+H]
+ 299.1178; found, 299.1192; IR (film): 2962, 2884, 2109, 1704, 1483, 1457, 1413, 1377, 1332, 1270, 1239, 1221, 1167, 1137, 1119, 1084, 1065, 1041, 983, 943, 923, 873, 855, 839, 807, 782, 758, 675, 634, 619, 553, 534, 495, 458 cm
-1.
27. House vacuum was used (130 mmHg) for this purpose.
28. Number of moles of
azidoacetylsultam 3 in the crude is calculated based on the purity percentage obtained from the quantitative NMR data.
29. The crude product was added as a solid to the reaction mixture. Larger pieces were broken gently with a glass rod before addition to the reaction.
30. HPLC grade
MeOH (certified ACS grade) was purchased from Fisher Scientific and used as received.
31.
Hydrochloric acid (36.5 to 38.0% (w/w), certified ACS grade) was purchased from Fisher Scientific and used as received.
32.
Palladium on carbon (10 wt%) was purchased from Sigma-Aldrich and used as received. The checkers purchased this catalyst from TCI.
33. The hydrogenolysis reaction using
palladium on carbon (10 wt%) creates a significant fire hazard if the activated catalyst is allowed to dry. Hence,
palladium on carbon was added to the reaction mixture mixed with water as a slurry to minimize the pyrophoricity.
34. During hydrogenolysis of azides, gaseous nitrogen is produced, hence every time before changing the
hydrogen balloon, a cycle of evacuating and back-filling was performed.
35. After 24 h, TLC analysis indicated that a substantial amount starting material was still present. Hence another portion of 10 wt%
Pd-C (2.50 g) was added following the described procedure.
36. The checkers observed remaining azide until the total reaction time had reached 40 h. The reaction was monitored by TLC, developed using (3:1, v/v)
hexanes-
ethyl acetate as the mobile phase and visualized using ninhydrin stain (Figure 13). The starting material forms a pink spot and the product forms a yellow spot upon heating with the stain. The R
f value of the starting material
1 was 0.41. The product doesn't move with the solvent and remains at the origin.
Figure 13. TLC analysis of the reaction mixture at 36 h (photo provided by submitter)
37. Celite 545 filtering aid (neutral or acidic - basic Celite leads to partial decomposition of the amine) was purchased form Fisher Scientific and used as received.
38. Care was taken to always maintain the solvent level above the Celite bed. Passing air through
Pd-C, trapped on the Celite bed can creates a significant fire hazard, especially in the presence of
methanol.
39. While concentrating, the product turned into a highly viscous liquid and then crystallized. After 24 h under 0.1 mmHg pressure, the large crystals were crushed using a spatula to facilitate the drying.
40. The submitters obtained 39.1 g of crude hydrochloride.
41.
Glycylsultam in the free amine form is highly reactive. In solution it can react with itself and undergo nucleophile-induced deacylation to give back the parent sultam
1, which is indicative of this decomposition pathway. Hence, trapping the amine as an acid salt is important, where in this case it is converted into the stable hydrochloride salt. The salt can be stored at -20 ℃ for an extended period without decomposition, and free amine can be regenerated easily by an aq
NaHCO3 neutralization.
42. Due to the excess amount of
hydrochloric acid that was added to the reaction mixture during the hydrogenolysis, the actual weight of the ammonium salt
5 is higher than the theoretical value. Hence, the entire crude ammonium salt product obtained from the previous step was finely crushed and homogenized before weighing out 5.00 g for the aq.
NaHCO3 neutralization. For the yield determination, a calculation was performed to determine the amount of free
glycylsultam 4 that can be obtained from the entire batch
5, based on the value received for a 5.00 g sample (
Note 46). The submitters used 5.00 g of crude material.
43. MColorpHast pH test strips (pH 6.5-10 and pH 5.2-7.2) were purchased from MilliporeSigma.
44. Due to the reactivity of the free amine, care was taken to perform the neutralization at 0 ℃ and to maintain the water bath temperature at 15-19 ℃ while concentrating the product under vacuum. When the neutralization and concentration were performed at higher temperatures, the product was observed to usually contain 3-5% of parent sultam
1 formed
via deacylation.
45. Completion of the neutralization was confirmed by monitoring the pH of the reaction mixture. If needed, the product
4 can be visualized by TLC developed using (9:1, v/v)
dichloromethane-
methanol as the mobile phase and visualized using ninhydrin stain. The product forms a yellow spot with a R
f value of 0.40 (Figure 14).
Figure 14. TLC analysis of the Oppolzer's glycylsultam 4 (photo provided by submitter)
46.
Glycylsultam 4 has the following characteristics (data provided by checkers): mp 118 - 121 ℃; [α]
58923 +115.6 (c 1.00, CHCl
3);
1H NMR
pdf (400 MHz, CDCl
3) δ: 3.93-3.84 (m, 2H), 3.76 (d,
J = 18.1 Hz, 1H), 3.49 (d,
J = 13.8 Hz, 1H), 3.43 (d,
J = 13.8 Hz, 1H), 2.16 (dd,
J = 12.4, 4.9 Hz, 1H), 2.12 - 2.04 (m, 1H), 1.96 - 1.83 (m, 3H), 1.46 (d,
J = 8.6 Hz, 2H), 1.42 (d,
J = 8.9 Hz, 1H), 1.36 (t,
J = 9.4 Hz, 1H), 1.14 (s, 3H), 0.97 (s, 3H);
13C NMR
pdf (176 MHz, CDCl
3) δ: 173.1, 65.3, 52.9, 49.3, 48.0, 45.6, 44.8, 38.4, 33.0, 26.6, 20.9, 20.0; IR (film): 3412, 3353, 2970, 2920, 2887, 2360, 2176, 1688, 1541, 1457, 1420, 1377, 1317, 1269, 1235, 1218, 1164, 1130, 1113, 1083, 1061, 1041, 981, 940, 910, 874, 815, 766 cm
-1. Purity of (
4) was assessed to be 99% based on qNMR
pdf analysis using
1,3,5-trimethoxybenzene as an internal standard. The checkers performed a second run on half scale, performing isolation on 1 g of crude material. This extraction yielded 0.58 g of product (99% purity) for a total yield of 83%.
47. The submitters reported the following result (based on 5.00 g of crude hydrochloride): 3.19 g, 89% yield, 98% purity.
Following are the calculations to determine the amount of free
glycylsultam 4 present in the entire ammonium salt
5 batch (39.1 g), based on the neutralization data received for 5.00 g sample and the percent yield.
Figure 15. Asymmetric multicomponent [C+NC+CC] synthesis of pyrrolidines. (XR and XS = antipodes of Oppolzer's camphorsultam)
Table 1. Application of the asymmetric [C+NC+CC] reaction to the synthesis of pyrrolidine containing natural products and drugs
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved