Org. Synth. 2021, 98, 509-530
DOI: 10.15227/orgsyn.098.0509
Preparation of 2-(Triethylsilyl)cyclohex-1-en-1-yl Trifluoromethanesulfonate as a Precursor to Cyclohexyne
Submitted by Kazuki Inoue, Kengo Inoue, Atsunori Mori, and Kentaro Okano*
1Checked by Zhixun Wang and Kevin Campos
1. Procedure (Note 1)
A. 2-(Triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (4). An oven-dried 1-L three-necked flask equipped with two 125-mL pressure-equalizing dropping funnels with a rubber septum, a Teflon-coated magnetic stir bar (4.5 x 1.5 cm), and a rubber septum inserted with a nitrogen gas inlet and a thermocouple, is charged with t-BuOK (7.01 g, 62.5 mmol, 2.5 equiv) (Note 2) and anhydrous n-hexane (100 mL) (Note 3). A solution of LDA (1.0 M in THF/hexanes, 62.5 mL, 63 mmol, 2.5 equiv) (Note 4) is added by cannula to the one of the dropping funnels, and this LDA solution is added dropwise over 5 min to the round-bottomed flask. The resulting mixture is stirred at ambient temperature for 30 min (Figure 1).
Figure 1. A) LDA being transferred into the dropping funnel via cannula; B) reaction mixture after addition of LDA and stirring at ambient temperature for 30 min (photos provided by checkers)
(Cyclohex-1-en-1-yloxy)triethylsilane (1) (5.57 g of 95% purity, 25.0 mmol, 1 equiv) (Note 5) is added to the reaction mixture via a syringe through the septum over 1 min (Figure 2). After stirring at ambient temperature for 1.0-2.5 h (Note 6), distilled H2O (675 μL, 37.5 mmol, 1.5 equiv) (Note 7) in anhydrous THF (100 mL) (Note 8) is added to the reaction mixture over 20 min (Note 9). After stirring at ambient temperature for 1 h, the reaction mixture is cooled to -78 °C with a dry ice-acetone bath. Comins' reagent (19.6 g, 50.0 mmol, 2.0 equiv) (Note 10) in anhydrous THF (75 mL) is added to the other pressure-equalizing dropping funnel and this solution is added dropwise to the round-bottomed flask over a 10-min period (Note 11) (Figure 3).

Figure 2. A) Reaction mixture after addition of (cyclohex-1-en-1-yloxy)triethylsilane (1) and stirring at ambient temperature for 1 h; B) reaction mixture after addition of water in THF and stirring at ambient temperature for 1 h (photos provided by checkers)
Figure 3. A) Reaction mixture after addition of Comins' reagent solution in THF at -78 °C; B) reaction mixture warming to ambient temperature (photos provided by checkers)
The solution is allowed to stir for another 10 min and then the cooling bath is removed. The reaction mixture warms to ambient temperature and is allowed to stir for 1 h (Note 12). The reaction is quenched with water (100 mL), which is added in one portion. The reaction mixture is transferred into a 1-L separatory funnel and extracted with n-hexane (3 x 100 mL). The combined organic extracts are washed with sat. aq. NaCl solution (1 x 150 mL), dried over Na2SO4 (25 g), and filtered. The filtrate is concentrated on a rotary evaporator under reduced pressure (15 °C, 10 mmHg), and the residue is dried in vacuo to afford 11.0-11.8 g of a crude product as a brown oil, which is purified by column chromatography on silica gel (elution: hexanes/Et2O = 19:1) (Notes 13 and 14) to provide 2-(triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (4) as a colorless oil (7.00 g, 81%) (Note 15) (Figure 4).

Figure 4. 2-(Triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (4) (photo provided by checkers)
B. 9,10-Diphenyl-1,2,3,4,9,10-hexahydro-9,10-epoxyanthracene (7). A 500-mL one-necked round-bottomed flask equipped with a Teflon-coated magnetic stir bar (4.5 x 1.5 cm) is charged with 1,3-diphenylisobenzofuran (6) (4.87 g, 18.0 mmol, 1.5 equiv) (Note 16), 2-(triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (4) (4.23 g of 98% purity, 12.0 mmol, 1 equiv), and THF (165 mL). A thermocouple is inserted into the reaction mixture. Tetrabutylammonium fluoride (1.0 M in THF, 18.0 mL, 18 mmol, 1.5 equiv) (Note 17) is added to the flask during 15 min via a syringe (Note 18), and after 1 h the thermocouple is removed (Figure 5).
Figure 5. Reaction mixture after addition of tetrabutylammonium fluoride (photo provided by submitters)
After stirring at ambient temperature for 2-4 h (Note 19), the reaction is quenched with water (120 mL). The reaction mixture is transferred into a 1-L separatory funnel and extracted with Et2O (2 x 120 mL). The combined organic extracts are washed with sat. aq. NaCl solution (1 x 80 mL), dried over Na2SO4 (10 g), and filtered. The filtrate is concentrated on a rotary evaporator under reduced pressure (35 °C, 10 mmHg), and the residue is dried in vacuo to afford 10.3-12.2 g of a crude product as a yellow solid, which is purified by column chromatography on silica gel (Note 20) to provide 9,10-diphenyl-1,2,3,4,9,10-hexahydro-9,10-epoxyanthracene (7) as a pale yellowish green solid (4.05 g, 96%, uncorrected for purity).
The resulting solid is recrystallized from boiling ethanol (200 mL) in a 500-mL one-necked round-bottomed flask equipped with a reflux condenser and a Teflon-coated magnetic stir bar (4.5 x 1.5 cm). Crystallization is allowed to occur at ambient temperature over 2 h, then at 0 °C for 30 min. The crystals are collected by filtration and washed with cold (0 °C) ethanol (30 mL) to provide the title compound 7 as colorless crystals (3.55 g, 84%) (Note 21) (Figure 6).
Figure 6. A) Filtration of product 7; B) appearance of recrystallized product 7 (photos provided by checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
lithium diisopropylamide,
potassium tert-butoxide,
n-hexane,
tetrahydrofuran,
Comins' reagent,
sodium chloride, anhydrous
sodium sulfate,
diethyl ether,
1,3-diphenylisobenzofuran,
tetrabutylammonium fluoride,
ethyl acetate, silica gel,
ethanol,
deuterated chloroform, and
1,3,5-trimethoxybenzene, as well as the proper procedures for setting up experimental operations.
2.
t-BuOK (>95.0%) was purchased from Tokyo Kasei Kogyo Co., Inc. by submitters and used as received without further purification.
t-BuOK (>98%) was purchased from Sigma-Aldrich Co. by checkers and used as received without further purification.
3. Anhydrous
n-hexane (>96.0%,
water content: < 30 ppm) was purchased from Nacalai Tesque Co., Inc. by submitters and used as received without further purification. Anhydrous
n-hexane (95%,
water content: < 10 ppm) was purchased from Sigma-Aldrich Co. by checkers and used as received without further purification.
4.
LDA (1.0 M in
THF/hexanes) was purchased from Sigma-Aldrich Co. by both submitters and checkers, and the solution was used as received.
5. The starting
(cyclohex-1-en-1-yloxy)triethylsilane (
1) was prepared as reported in
Org. Synth. 2021,
98, 407-429.
6. The reaction typically requires 1 h to consume all the silyl enol ether
1 and is monitored by TLC analysis on Merck silica gel 60 F
254 plates developing with hexanes/
ethyl acetate (9:1). The R
f values of
(cyclohex-1-en-1-yloxy)triethylsilane (
1) and 2-(triethylsilyl)cyclohexan-1-one are 0.86 and 0.45, respectively (stained with an
ethanol solution of
p-anisaldehyde). After dipping the TLC plate into the solution, the chromatogram is heated to reveal the stained compounds (Figure 7).
Figure 7. TLC of (cyclohex-1-en-1-yloxy)triethylsilane (1, left), reaction mixture after addition of 1 and stirring for 1 h (right), and their combination spot (middle). Eluent: hexanes/ethyl acetate (9:1, v/v) (photo provided by checkers)
7. Distilled
water was purchased from Nacalai Tesque Co., Inc. by submitters and used as received without further purification.
water (UHPLC-MS grade) was purchased from Thermo Fisher Scientific Inc. by checkers and used as received without further purification.
8.
THF (>99.5%,
water content: < 10 ppm) was purchased from Kanto Chemical Co., Inc. by submitters and further dried by passing through a solvent purification system (Glass Contour) prior to use. Anhydrous
THF (>99.9%, inhibitor-free,
water content: < 20 ppm) was purchased from Sigma-Aldrich Co. by checkers and used as received without further purification.
9. An exotherm (from 22 to 29 °C) was observed during the 20 min addition time at the reported scale. A larger scale reaction than that described in this procedure may require a longer addition time to control the exotherm.
10.
Comins' reagent (>99.0%) was purchased from Oakwood Products, Inc. by both submitters and checkers, and used as received without further purification.
11. An exotherm (from -72 to -65 °C) was observed during the 20 min addition time at the reported scale. A larger scale reaction than that described in this procedure may require a longer addition time to control the exotherm.
12. The reaction is monitored by TLC analysis on Merck silica gel 60 F
254 plates developing with hexanes/
ethyl acetate (9:1). The R
f values of 2-(triethylsilyl)cyclohexan-1-one and
2-(triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (
4) are 0.45 and 0.82, respectively (stained with an
ethanol solution of
p-anisaldehyde). After dipping the TLC plate into the solution, the chromatogram is heated to reveal the stained compounds (Figure 8).
Figure 8. TLC of reaction mixture before addition of Comins' reagent (left), reaction mixture after addition of Comins' reagent and warming up to rt (right), and their combination spot (middle). Eluent: hexanes/ethyl acetate (9:1, v/v) (photo provided by checkers)
13. Silica gel (360 g, spherical, 100 μm, pH 6-8, purchased from TCI Chemicals by both submitters and checkers) and 0.9 L hexanes/Et
2O (19:1) were charged into a 1-L bottle, and shaken up to ensure mixing. The resulting silica gel slurry was loaded into the column (d = 6.5 cm, h = 20 cm). The crude product was suspended in 10 mL hexanes/Et
2O (19:1), and loaded onto the column, after which another 10 mL hexanes/Et
2O (19:1) was added to the flask to ensure a complete transfer. The column was eluted with hexanes/Et
2O (19:1). A total of 40 fractions (25 mL) were collected (total eluting time 15-20 min). Analysis by TLC (
Note 12) revealed that the product was obtained in fractions 19-28, which were combined and concentrated on a rotary evaporator under reduced pressure (40 °C, 100 to 7 mmHg) (Figure 9).
Figure 9. TLC of the fractions from flash column chromatography. Eluent: hexanes/Et2O (19:1, v/v) (photo provided by checkers)
14. Both submitters and checkers observed that
2-(triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (
4) undergoes decomposition on silica gel during column chromatography. Extensive optimization indicates that silica gel brands, silica gel/crude product ratio, and hexanes/Et
2O ratio in eluent (as shown in
Note 13) are crucial to deliver satisfactory yield and purity.
15.
2-(Triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (
4): colorless oil; R
f = 0.79 (hexanes/Et
2O = 19:1); Merck silica gel 60 F
254 plates (stained with an
ethanol solution of
p-anisaldehyde). After dipping the TLC plate into the
p-anisaldehyde solution, the chromatogram is stained by heating; IR (film, ATR): 2953, 1648, 1408, 1201, 1143, 986, 884, 850, 722 cm
-1;
1H NMR
pdf (500 MHz, CDCl
3) δ: 0.73 (q,
J = 7.9 Hz, 6H), 0.95 (t,
J = 7.9 Hz, 9H), 1.51 - 1.63 (m, 2H), 1.69 - 1.81 (m, 2H), 2.19 (tt,
J = 6.0, 2.5 Hz, 2H), 2.42 (tt,
J = 6.4, 2.4 Hz, 2H);
13C NMR
pdf (126 MHz, CDCl
3) δ: 2.9, 7.6, 21.9, 23.2, 28.5, 28.9, 118.4 (q,
1JC-F = 320 Hz), 125.6, 155.2;
19F NMR
pdf (471 MHz, CDCl
3) δ: -74.77; The purity of the sample was determined to be 98% by
1H qNMR
pdf using 37.42 mg of
1,3,5-trimethoxybenzene (Note 22) as an internal standard and 40.90 mg of product
4. Based on the purity, 6.86 g (79%) of the product was prepared. A second reaction on the identical scale provided 7.19 g of the product with 94% purity.
16.
1,3-Diphenylisobenzofuran (
6) (>97.0%) was purchased from Tokyo Kasei Kogyo Co., Inc. by submitters and used as received without further purification.
1,3-Diphenylisobenzofuran (
6) (97%) was purchased from Sigma-Aldrich Co. by checkers and used as received without further purification.
17.
Tetrabutylammonium fluoride (1.0 M in
THF) was purchased from Sigma-Aldrich Co. by both submitters and checkers, and the solution was used as received without further purification.
18. A small exotherm (+8 °C) was observed during 15 min addition time at current scale.
19. The reaction typically requires 2-4 h to consume all the
2-(triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (
4) and is monitored by TLC analysis on Merck silica gel 60 F
254 plates developing with
n-hexane/CH
2Cl
2 (9:1). The R
f values of
2-(triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (
4),
1,3-diphenylisobenzofuran (
6), and
9,10-diphenyl-1,2,3,4,9,10-hexahydro-9,10-epoxyanthracene (
7) are 0.68, 0.53, and 0.36, respectively (stained with an
ethanol solution of
p-anisaldehyde). After dipping the TLC plate to the solution, the chromatogram is stained by heating (Figure 10).
Figure 10. Left: TLC of 2-(triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (A) and the reaction mixture (B) (stained with an ethanol solution of p-anisaldehyde). Right: TLC of 2-(triethylsilyl)cyclohex-1-en-1-yl trifluoromethanesulfonate (A) and the reaction mixture (B) (visualized by UV lamp) (photos provided by submitters)
20. The crude product was dissolved in CH
2Cl
2 (40 mL) and 30-gram silica gel was added. The resulting slurry was evaporated to dryness on a rotary evaporator (35 °C, 100 to 10 mmHg). The crude product mixture was charged onto a RediSep RF column (diameter = 3.70 cm, height = 21.8 cm, 120-gram silica gel). The column was eluted with linear gradient from 0 to 25% CH
2Cl
2 in hexanes on a ISCO CombiFlash system, and 50-mL fractions were collected. Fractions 14-27 were combined and concentrated on a rotary evaporator under reduced pressure (40 °C, 100 to 10 mmHg).
21.
9,10-Diphenyl-1,2,3,4,9,10-hexahydro-9,10-epoxyanthracene (
7): color-less crystals; R
f = 0.36 (
n-hexane/CH
2Cl
2 = 9:1); Merck silica gel 60 F
254 plates (stained with an
ethanol solution of
p-anisaldehyde). After dipping the TLC plate to the
p-anisaldehyde solution, the chromatogram is stained by heating; mp 171-173 °C (
ethanol); IR (film): 2920, 2853, 2826, 1601, 1446, 1307, 996, 742, 699 cm
-1;
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.40 - 1.51 (m, 2H), 1.55 - 1.69 (m, 2H), 1.99 - 2.16 (m, 2H), 2.22 - 2.38 (m, 2H), 6.99 (dd,
J = 5.2, 3.0 Hz, 2H), 7.24 (dd,
J = 5.2, 3.0 Hz, 2H), 7.40 - 7.46 (m, 2H), 7.49 - 7.57 (m, 4H), 7.73 - 7.80 (m, 4H);
13C NMR
pdf (126 MHz, CDCl
3) δ: 22.4, 23.4, 92.3, 119.0, 124.6, 126.3, 127.7, 128.4, 135.5, 150.2, 151.8; The purity of the sample was determined to be >99% by
1H qNMR
pdf using 39.24 mg of
1,3,5-trimethoxybenzene (Note 22) as an internal standard and 47.23 mg of product
7. A second reaction on the identical scale provided 3.99 g of crude product, which provided 3.48 g (83%, uncorrected for purity) of the product after recrystallization.
22.
1,3,5-Trimethoxybenzene (>99%) was purchased from Sigma-Aldrich Co. and used as received.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Strained molecules have attracted a great deal of attention because of their high reactivities.
Cyclohexyne (
5), in which a triple bond is present in a six-membered ring, is a short-lived intermediate and has been extensively investigated to construct the various molecular frameworks,
2 despite the limited number of reports compared to aryne
3 (Scheme 1).
Scheme 1. Representative examples in constructing molecular frameworks using cyclohexyne (5)
Similar to the generation of 1,2-cyclohexadiene (
8),
cyclohexyne (
5) can be generated from silylated cyclohexenyl triflate
9 with the aid of fluoride ion reported by Guitián
2b (Scheme 2). Compared to other methods for generating cyclohexynes,
4 this fluoride ion-promoted conditions show the superior functional group compatibility. The only drawback of this method is the multi-step preparation of the silylated cyclohexenyl triflate
9. The synthesis started with bromination of cyclohexenone (
10), whose carbonyl group was protected as its acetal to provide
11. The alkenyl bromide underwent halogen-lithium exchange followed by silylation. Aqueous workup gave 2-silylcyclohexenone
12. The use of L-Selectride facilitated 1,4-reduction, and the resultant lithium enolate
13 was treated with PhNTf
2 to yield the silylated cyclohexenyl triflate
9. In 2006, Peña reported generation of 1,2-cyclohexadiene (
8) from silylated cyclohexenyl triflate
14 under the same reaction conditions. They first prepared 2-silylcyclohexanone
15 by protonation of lithium enolate
13. Subsequent treatment with
LDA led to the formation of trisubstituted lithium enolate
16 through kinetic deprotonation. The following triflation provided cyclohexenyl triflate
14 that is the precursor for 1,2-cyclohexadiene (
8). Most of elaboration was dedicated to the introduction of the trimethylsilyl group, which is the key to shorten the synthesis of both silylated cyclohexenyl triflates.
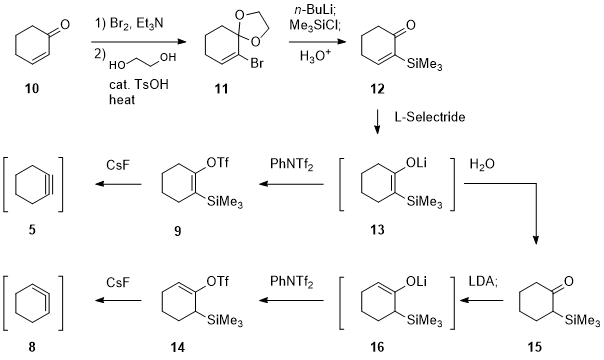
Scheme 2. Guitián and Peña's synthesis of the silylated cyclohexenyl triflates for cyclohexyne (5) and 1,2-cyclohexadiene (8)
As reported in our synthesis of 1,2-cyclohexadiene precursor
17,
5 transfer of the triethylsilyl (TES) group in silyl enol ether
1, which is easily prepared from cyclohexanone (
18), is the powerful method to realize the facile synthesis of compound
17 (Scheme 3). Apart from the Guitián and Peña's method
2b,6a,6b and recently reported Garg's method,
6c,6d we first generate thermodynamically unstable trisubstituted silylated lithium enolate
19, by rearrangement of allyllithium
20. Subsequent triflation takes place smoothly with
Comins' reagent7 to provide 1,2-cyclohexadiene precursor
17 in 80-84% yield on a 7-gram scale from TES enol ether
1 in a single flask.
5c We then devised an idea that isomerization of trisubstituted silylated lithium enolate
19 via 2-silylcyclohexanone
21 gives tetrasubstituted silylated lithium enolate
2, which should be transformed to the corresponding
cyclohexyne precursor
4. We first performed the isomerization simply by prolonging reaction time and found that
i-Pr
2NH, the conjugate acid of
LDA, promoted the desired isomerization.
5a We have also examined a variety of proton sources and found that
water was the best additive to facilitate this isomerization of the double bond. The amount of
water was carefully optimized; 1.5 equivalents of
water were found to be best for the high-yielding process.
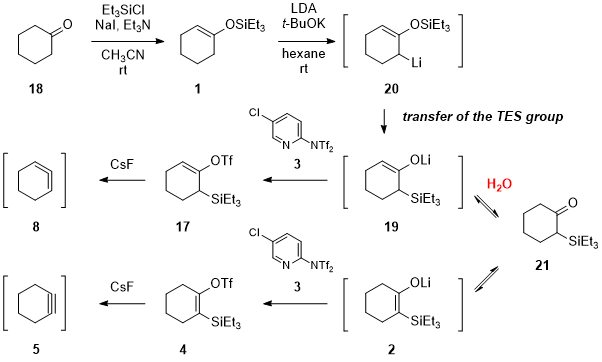
Scheme 3. Our synthetic strategy for the divergent synthesis of the silylated cyclohexenyl triflates for cyclohexyne (5) and 1,2-cyclohexadiene (8)
The amount of
water was crucial for this isomerization (Table 1). In the absence of
water, 1,2-cyclohexadiene precursor
17 was exclusively obtained (entry 1).
5c As the amount of the
water increased, the yields of
cyclohexyne precursor
4 were improved; 1.5 equivalents proved most effective for the selective synthesis of silylated cyclohexenyl triflate
4 (entries 2-4). The excess amount of
water resulted in significant reduction of the yields of compound
4 with concomitant formation of 2-silylcyclohexanone
21 by protonation (entries 5 and 6).
Table 1. Effects of water on the yields of the silylated cyclohexenyl triflates
The established optimal conditions could be applied to various silylated cyclohexenyl triflates (Table 2). In addition to the TES enol ether, the TBS ether underwent the transfer of the silyl group and subsequent isomerization, which was then treated with Comins' reagent to give the desired product in moderate yield. The TIPS enol ether and TBDPS enol ether were converted to the corresponding products, albeit in low yields. The TES enol ether bearing the tert-butyl group at position 4 was transformed to the product in 59% yield as a sole regioisomer, whereas the TES enol ether bearing the tert-butyl group at position 3 was converted to the product in 44% yield.
Table 2. Scope of the reaction
In summary, the described protocol provides a reliable method for the synthesis of both silylated cyclohexenyl triflates that are precursors for cyclohexyne and 1,2-cyclohexadiene on multigram scales. The presented method is operationally simple and robust, and the starting cyclohexanone is widely available. This method can be applied to several silyl groups except for the trimethylsilyl group, which would promote the research on these reactive and strained synthetic intermediates for the synthesis of natural products, medicines, and functional organic materials.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
t-BuOK: 2-Propanol, 2-methyl-, potassium salt (1:1); (865-47-4)
Hexane: hexane; (110-54-3)
LDA: 2-Propanamine, N-(1-methylethyl)-, lithium salt (1:1); (4111-54-0)
THF: Furan, tetrahydro-; (109-99-9)
Water; (7732-18-5)
Comins' reagent: Methanesulfonamide, N-(5-chloro-2-pyridinyl)-1,1,1-trifluoro-N-[(trifluoromethyl)sulfonyl]- (145100-51-2)
1,3-Diphenylisobenzofuran: Isobenzofuran, 1,3-diphenyl-; (6) (5471-63-6)
Tetrabutylammonium fluoride: 1-Butanaminium, N,N,N-tributyl-, fluoride (1:1); (429-41-4)

|
Kazuki Inoue was born in Osaka in 1994. He received his B.S. in 2017 and M.S. in 2019 from Kobe University under the direction of Professors Atsunori Mori and Kentaro Okano. During his M.S. studies, he worked on generation of strained cycloalkynes and its application to the synthesis of functionalized poly(cycloalkyne)s. Currently, he works for Sumitomo Bakelite Co., Ltd. |

|
Kengo Inoue was born in Fukuoka in 1997. He received his B.S. in 2020 from Kobe University under the direction of Professors Atsunori Mori and Kentaro Okano. During his M.S. studies, he is working on selective trapping of short-lived heteroaryllithiums in halogen dance and its application to the synthesis of multiply substituted heteroaromatic compounds. |

|
Atsunori Mori was born in Aichi in 1959. He received B.S. (1982), M.S. (1984), and Ph.D. (1987) degrees from Nagoya University under the direction of Professor Hisashi Yamamoto. After postdoctoral study in U.C. Berkeley with Peter Vollhardt (1987-1988), he started his academic career as an Assistant Professor at the University of Tokyo (1988) and Japan Advanced Institute of Science and Technology (1993). He was appointed as an Associate Professor in 1995 at Chemical Resources Laboratory of Tokyo Institute of Technology. Since 2005, he has been Professor at the Graduate School of Engineering, Kobe University. |

|
Kentaro Okano was born in Tokyo in 1979. He received his B.S. in 2003 from Kyoto University under the supervision of Professor Tamejiro Hiyama. He then moved to the laboratories of Professor Tohru Fukuyama at the University of Tokyo. In 2007, he joined the faculty at Tohoku University in Professor Hidetoshi Tokuyama's group. In 2014, he visited Professor Amir Hoveyda's laboratories at Boston College as a visiting researcher. In 2015, he moved to Kobe University, where he is currently Associate Professor of Department of Chemical Science and Engineering. His current research interest is natural product synthesis based on the development of new synthetic methodologies. |

|
Zhixun Wang joined the Process Research Department of Merck & Co., Inc. in 2019. His research focuses on synthesize therapeutic candidates for large-scale production. He received his B. S. degree from Fudan University, and then obtained his Ph.D. under the supervision of Professor Liming Zhang at University of California, Santa Barbara with a research focus on homogeneous gold catalysis. Zhixun then moved to New Haven, CT, where he did postdoctoral work on synthesis of pleuromutilin analogs and structure elucidation of colibactin with Professor Seth Herzon in Yale University. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved