Diphenylprolinol silyl ether
3 (Figure 1) is a versatile organocatalyst.
2 In 2005, our group reported the asymmetric Michael reaction of aldehydes and nitroalkenes catalyzed by this catalyst
3.
3 In 2011, we found that the reaction is greatly accelerated by
p-nitrophenol.
4 The procedure based on this finding is described in our article of 2017, published in
Organic Syntheses.5Figure 1. Representative organocatalysts
In 2005, Jørgensen and coworkers reported the sulfenylation of aldehydes using diarylprolinol with trifluoromethyl substituents on the phenyl group
4.
6 In both Michael and sulfenylation reactions, an enamine, generated from the aldehyde and the catalyst, is a key intermediate (eq. 1). As the enamine is a reactive nucleophile, it reacts with electrophiles to afford α-functionalized aldehydes with excellent enantioselectivity. This type of catalyst also reacts with an α,β-unsaturated aldehyde to generate an iminium ion (eq. 2). The iminium ion is a reactive electrophile; it reacts with nucleophiles to afford β-functionalized aldehydes with excellent enantioselectivity.
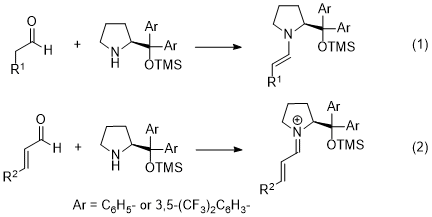
Here, there are two major catalysts: diphenylprolinol silyl ether
3, and diarylprolinol silyl ether with trifluoromethyl substituents
4 (Figure 1). The difference between these two catalysts is as follows.
7 As a trifluoromethyl substituent is a good electron-withdrawing group, the electron density on the nitrogen in catalyst
4 is lower than that on the nitrogen in catalyst
3. The latter is a nucleophilic catalyst and the HOMO level of the enamine generated from catalyst
3 is higher than that generated from catalyst
4. Thus, the reactivity of the enamine generated from
3 is higher than that of the enamine generated from
4. Regarding the iminium ion, on the other hand, the LUMO of the iminium ion generated from
4 is lower than that generated from
3, and the former iminium ion is more electrophilic and reactive.
The silyl group is essential for the high reactivity. In the reaction of diphenylprolinol, the reaction is slow. This is because diphenylprolinol reacts with an aldehyde to generate a stable N,O-acetal with low concentration of the reactive enamine (eq. 3). However, we found that diarylprolinol is effective for the cross-aldol reaction of two different aldehydes, in which the hydroxy group of the catalyst is essential for the activation of the electrophilic aldehydes (eq. 4).
8In both reactions involving the enamine and the iminium ion as an intermediate, excellent enantioselectivity is realized. This could be because of steric effects in the transition state (Figure 2). One of the enantiofaces of the enamine and the iminium ion is effectively shielded by the bulky diarylmethyl silyl ether moiety, facilitating approach of an electrophile or a nucleophile from the opposite side.
Figure 2. Effect of the silyl moiety
Regarding the silyl substituent, not only can a trimethylsilyl group be utilized, but other silyl groups, such as triethylsilyl,
tert-butyldimethylsilyl, triphenylsilyl, and triisopropylsilyl can be used, as well. In general, as the size of the silyl group increases, the generation of the enamine and the iminium ion from the corresponding aldehyde and α,β-unsaturated aldehyde, in the presence of catalyst, becomes slower because of steric hindrance, while the enantioselectivity generally increases.
9In our article of 2017, published in
Organic Syntheses,
5 we reported the asymmetric Michael reaction of propanal and nitrostyrene. Michael product was obtained in excellent yield with excellent diastereoselectivity and enantioselectivity. The reaction mechanism revealed that this is not a simple Michael reaction. As shown in Scheme 1,
4, 10 the enamine and nitroalkene react via [2+2] and [4+2] cycloaddition pathways to afford cyclobutane
5 and dihydro-oxazine
N-oxide
6, which are stable and can be isolated. Compounds
5 and
6 can be transformed into the Michael product by reaction with water, accelerated by acid, which is why a catalytic amount of acid accelerates the reaction.
Scheme 1. Reaction mechanism of the Michael reaction of propanal and nitrostyrene
Regarding the Michael reaction of aldehydes and nitroalkenes, an aldehyde lacking substitution at the α-position, such as acetaldehyde, can be successfully employed as a Michael donor, affording the product with excellent enantioselectivity (eq. 5).
11Moreover, not only nitroalkenes but also β-substituted α-nitroacrylates,
12 dicyanoalkenes,
13 and α-cyano α,β-unsaturated esters,
14 and vinyl sulfones
15 can be employed as useful Michael acceptors, using an aldehyde as a Michael donor (eq. 6). In all cases, excellent enantioselectivity is realized.
There are several applications of the asymmetric Michael reaction of aldehydes and nitroalkenes in the total synthesis of biologically-active molecules. Chemists at Novartis have reported the total synthesis of aliskiren (Rasilez), a novel renin inhibitor, by using the asymmetric Michael reaction of isovaleraldehyde and nitroethene, generated from 2-(benzoyloxy)-1-nitroethane
in situ, as one of the key steps (Scheme 2).
16Scheme 2. Michael reaction in the total synthesis of aliskiren (Rasilez) by Novartis
The Michael reaction of an α-alkoxy aldehyde and nitroalkene was successfully employed in the pot- and time-economical total synthesis of (-)-oseltamivir (Scheme 3) (one pot, 60 min).
17Scheme 3. Total synthesis of (-)-oseltamivir: one pot, 60 min
Diphenylprolinol silyl ether is a secondary amine catalyst with a bulky substituent, and its basicity is rather low. It activates an aldehyde and an α,β-unsaturated aldehyde selectively, but it has no effect in other reactions because it is a weak base. This catalyst is also effective in domino reactions. A domino reaction is a reaction involving two or more bond-forming transformations that take place under the same reaction conditions without the addition of further reagents and catalysts.
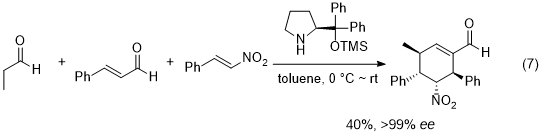
18 One of the early successful examples is Enders' domino Michael/Michael/aldol condensation reaction of an aldehyde, nitroalkene, and α,β-unsaturated aldehyde, affording substituted cyclohexene derivatives with excellent enantioselectivity (eq. 7).
19
The domino Michael/Henry reaction of succinaldehyde and a nitroalkene was catalyzed by diphenylprolinol silyl ether to afford a cyclopentane derivative (Scheme 4).
20 This is a formal [3+2] cycloaddition reaction, successfully used in the three-pot synthesis of prostaglandin E
1 (PGE
1) methyl ether,
21 and a short synthesis of beraprost.
22Scheme 4. Synthesis of PGE1 methyl ester
Diphenylprolinol silyl ether was also utilized in the total synthesis of a steroid. A domino Michael/intramolecular aldol reaction of nitroalkane
7 and an α,β-unsaturated aldehyde was catalyzed by diphenylprolinol silyl ether to afford a bicyclo[4.3.0]nonane derivative
8, which corresponds to the C and D rings of the steroid, with nearly enantiomerically pure form, with the generation of an all-carbon quaternary center (Scheme 5).
23 This compound
8 was efficiently converted into estradiol methyl ether.
Scheme 5. Synthesis of estradiol methyl ether
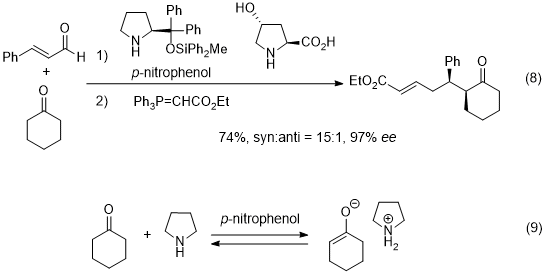
Recently, the Michael reaction of a ketone as a nucleophile was reported, in which use was made of two amine catalysts such as diphenylprolinol silyl ether and hydroxyproline (eq. 8).
24 In this reaction, the key nucleophile is an enolate, which is generated from a ketone in the presence of a weak acid and a base (eq. 9).
25
Based on this Michael reaction, a domino Michael/Michael reaction of an α,β-unsaturated aldehyde and ethyl 4-oxo-2-pentenoate (
9) was developed (Scheme 6).
26 The catalyst reacts with an α,β-unsaturated aldehyde to generate an iminium ion
10, which reacts with ketone
9 via a Michael reaction to afford an enamine
11. A diastereoselective second Michael reaction proceeds from
11 to afford a substituted cyclopentanone derivative with excellent diastereoselectivity. In this domino double Michael reaction, nearly enantiomerically pure product was obtained. This is because the chiral catalyst is involved in both steps and kinetic resolution occurs in the second Michael reaction. Using this reaction as a key step, Corey lactone was synthesized in a one-pot reaction.
Scheme 6. Synthesis of Corey lactone
In summary, diphenylprolinol silyl ether is an effective organocatalyst for both the enamine and the iminium ion as reactive intermediate. Using this catalyst, new asymmetric reactions have been reported-chiral complex structures are constructed by a domino reaction.
27 Several successful combinations of this organocatalyst and metal catalysts have been reported.
28 The combination of a photocatalyst and organocatalyst is a growing field.
29 Applications of the asymmetric reaction catalyzed by this catalyst are increasing-natural products and pharmaceuticals are effectively synthesized by this catalyst.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved