1,1-Difluoroallenes have two fluorine substituents, which are located on the cumulated diene substructure to decisively affect their reactivities. In addition to the reactions compiled in our previous review,
2 synthetic reactions of 1,1-difluoroallenes have been continuously investigated since the authors' publication in
Org. Synth. that described difluoroallene production by difluorovinylidenation of aldehydes and ketones.
3 As a result, the fluorine substituents allow bond-forming reactions of 1,1-difluoroallenes to proceed in α-, β-, or γ-selective fashions, all of which are illustrated below.
Bond Forming Reactions on the α-Carbon
As discussed in the original article,
3 an In(III) catalyst facilitates the domino Friedel-Crafts-type cyclization/ring expansion sequence of 1,1-difluoroallenes
1. Subsequent one-pot dehydrogenation leads to pinpoint-fluorinated polycyclic aromatic hydrocarbons
2 (F-PAHs, Scheme 1).
4 The domino reaction allows two carbon-carbon bond formations, first at the position α to the fluorine substituents and subsequently at the γ-position.
Scheme 1. In(III)-catalyzed synthesis of F-PAHs
Three applications were developed using the above domino reaction to construct extended π systems, namely, (i) tandem benzene ring construction,
5 (ii) π-extended aryne generation,
6 and (iii) benzene ring extension.
7 These applications resulted in the successful generation of difluorinated, monofluorinated, and fluorine-free π-extended molecular systems.
The first application of the domino cyclization/ring expansion sequence involves tandem benzene ring construction (Figure 1).
5 Bis(1,1-difluoroallene)
3 (a) and
4 (b), prepared from
m- and
p-xylenes via the corresponding dialdehydes, underwent the domino reaction in a tandem fashion to afford pinpoint-difluorinated dibenzoanthracene
5 and picene
6 in 76% and 75% yields, respectively. The physicochemical features of the synthesized F-PAHs were examined, specifically their solubility in organic solvents
8 and their potential as materials for electronic devices.
5Figure 1. Synthesis of pinpoint-difluorinated PAHs (F-PAHs)
Secondly, the synthesis of "half HBCs" was facilitated by π-extended aryne generation from 1,1-difluoroallenes (Scheme 2).
6 The structure of half HBCs is a half section of HBCs (hexabenzocoronenes), which are promising as materials for photovoltaic cells.
o-Bromo- or
o-iodofluoroarenes
7 were prepared by the In(III)-catalyzed domino reaction of 1,1-difluoroallenes involving halogenation of the C-In bond with
N-bromosuccinimide (NBS) or
N-iodosuccinimide (NIS).
4a Compounds
7 served as precursors for π-extended arynes
8 upon treatment with butyllithium, while 6-fluoro[4]helicene (not shown) also served as an aryne precursor via dehydrofluorination upon treatment with Me
2 (TMP)ZnLi (TMP, tetramethylpiperidino).
9 The produced arynes were subjected to the Diels-Alder reaction with diarylated isobenzofurans,
10 yielding cycloadducts (81-89% yields, not shown), which were easily transformed to half HBCs
9 by deoxygenative aromatization followed by aryl-aryl coupling in 78-96% yields (2 steps).
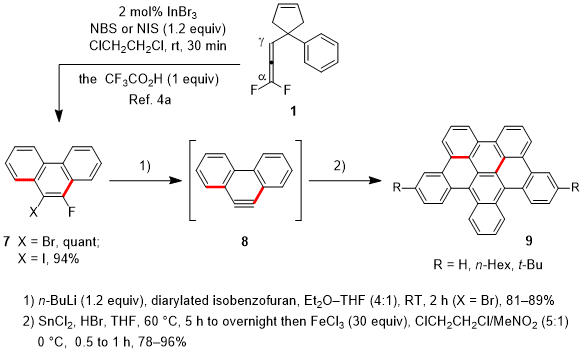
Scheme 2. Synthesis of half HBCs
Third, benzene ring extension was accomplished through the attachment of a fluorobenzo moiety to existing fluoroarenes.
7 Under microwave (MW) irradiation, commercially available 1-fluoronaphthalene was effectively cyanoethylated by aromatic nucleophilic substitution for fluorine (Figure 2). The half reduction of the nitrile gave aldehyde
10, whose difluorovinylidenation afforded the corresponding 1,1-difluoroallene
11. Next,
11 underwent Friedel-Crafts-type cyclization followed by dehydrofluorination to provide the benzene ring-extended fluorophenanthrene
12 in 94% yield (the first cycle). Application of second and third cycles similarly extended the π system until it eventually afforded pinpoint-fluorinated [5]phenacene (picene)
13 (Scheme 3). In addition to phenacenes with zig-zag benzene rings, such as
13, triphenylene
14 with a trigonal structure was produced by applying the benzene ring extension cycle to internally fluorinated arenes (Scheme 4).
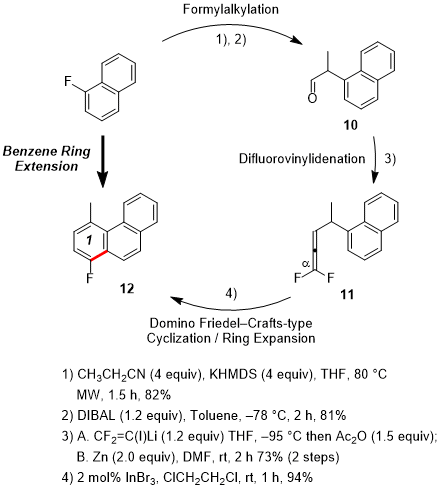
Figure 2. Benzene ring extension cycle
Scheme 3. Synthesis of fluorinated phenacenes
Scheme 4. Synthesis of fluorinated triphenylenes
Apart from cyclizations, 1,1-difluoroallenes also undergo α-selective addition of oxygen nucleophiles, such as phenols, carboxylic acids, and sulfonic acids, in the presence of an Au(I) or an Au(III) catalyst (Scheme 5).
11 Thus, using the aurated allylic CF
2 cations, the hard O-nucleophiles were regioselectively introduced to 1,1-difluoroallenes, yielding 1,1-difluoroallylic ethers and esters
15 in 74%-92% yields. In contrast, soft sulfur and nitrogen nucleophiles underwent γ-selective addition (vide infra: Scheme 9).
Scheme 5. Synthesis of 1,1-difluoroallylic ethers and esters
Bond Forming Reactions on the β-Carbon
Bond-forming reactions at the position β to the fluorine substituents were facilitated by a palladium catalyst involving π-allylpalladium(II) formation.
12 Difluoroallene
16 with a bromophenyl moiety was intramolecularly carbopalladated to form the π-allylpalladium(II) species with a six-membered ring structure, which in turn underwent β-hydrogen elimination, followed by isomerization to yield difluoromethylated naphthalene
17 in 60% yield (Scheme 6). The difluoromethyl group is a bioisostere of a hydroxy group and is attracting attention in the field of pharmaceuticals and agrochemicals as a hydrogen donor for hydrogen bonding while simultaneously exhibiting hydrophobicity.
Scheme 6. Synthesis of difluoromethylated naphthalenes
The intramolecular β-selective carbometallation was followed by intermolecular fluorometallation (cat. Pd(0)/PhI/AgF, Scheme 7),
13 where the generation of vinylsilver(I) was proposed to participate in Pd(0)-catalyzed coupling, leading to (trifluoromethyl)alkene
18 in 65% yield. A rhodium(I) catalyst bearing a nitrogen ligand facilitated C-S bond formation in a β-selective fashion (Scheme 8), while Rh(I) with a phosphine ligand promoted γ-addition (vide infra: Scheme 10).
14Scheme 7. Synthesis of arylated (trifluoromethyl)alkenes
Scheme 8. Synthesis of sulfanylated (difluoromethyl)alkenes
Bond Forming Reactions on the γ-Carbon
Organocopper(I) reagents promote γ-selective bond-forming reactions in almost all cases.
15 3-Monosubstituted 1,1-difluoroallene
19 reacted with ethylcopper(I) to afford the corresponding addition product, γ-branched 1,1-difluoro-1-alkene
20 (E = H), in 95% yield via protonolysis of the 2,2-difluorovinylcopper(I) intermediate (Figure 3).
16 When quenched by electrophiles, such as halogenating agents and halostannanes, β-functionalized 1,1-difluoro-1-alkenes
21 and
22, respectively (66%-84% yields). The 2,2-difluorovinylcopper(I) intermediates enabled Pd(0)-catalyzed coupling with iodobenzene, yielding a three-component coupling product
23 in 90% yield.
Figure 3. Synthesis of γ-branched 1,1-difluoro-1-alkenes
B
2pin
2 and PhMe
2SiBpin with a Cu(I)-catalyst promoted γ-selective borylation and silylation of 1,1-difluoroallenes, which afforded 3,3-difluoroallylboronate
24 and silane
25 in 86% yields, respectively (Figure 4).
17 The formed difluoroallylboronates reacted with aldehydes to provide 2,2-difluorohomoallylic alcohols (not shown).
Figure 4. Synthesis of 3,3-difluoroallylic boronates and silanes
As well as undergoing alkylation, borylation, and silylation reactions, 1,1-difluoroallenes underwent the γ-selective addition of benzamide and thiophenol in the presence of an Au(I) [or an Au(III)] catalyst through cationic intermediates (Scheme 9, see also: Scheme 5).
11 3,3-Difluoroallylic amine
26 and thioether
27 were synthesized in 81% and 72% yields, respectively. The use of a Rh(I) catalyst with a chiral phosphine ligand aided in the synthesis of chiral 3,3-difluoroallylic thioether
28 via γ-selective C-S bond formation (Scheme 10, see also: Scheme 8).
14Scheme 9. Synthesis of 3,3-difluoroallylic amines and thioethers
Scheme 10. Synthesis of chiral 3,3-difluoroallylic thioethers
The related monofluoroallenes underwent γ-selective intramolecular C-O and C-N bond formations in the presence of an Ag(I) catalyst (Scheme 11). Ring-fluorinated heterocycles, dihydropyran
29, and tetrahydropyridine
30, were obtained in 54% and 52% yields, respectively.
18Scheme 11. Synthesis of heterocyclic fluoroalkenes
In summary, since our report on the synthesis of 1,1-difluoroallenes by carbonyl difluorovinylidenation, the reactions of 1,1-difluoroallenes have been steadily investigated and their unique reactivities have been revealed. Now, regioselective bond-forming reactions in 1,1-difluoroallenes can be successfully effected at each of the three, α-, β-, and γ-positions with the aid of metals, such as In(III), Au(I), Au(III), Pd(0), Cu(I), and Ag(I), which enables the synthesis of fluorinated and fluorine-free cyclic and acyclic molecules. As a result, 1,1-difluoroallenes are extremely adaptable synthetic building blocks. Despite these advances in ionic reactions, further research on their behavior under radical conditions and in electrocyclization processes is still required.
19
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved