Checked by Tufan K. Mukhopadhyay and Dirk Trauner
1. Procedure (Note 1)
B. (3aR,6aS)-2-Hydroxy-3,3a,6,6a-tetrahydro-2H-cyclopenta[b]furan-5-carbaldehyde (2). To a 2 L round-bottomed flask equipped with a 4.0 cm magnetic stirrer bar is added freshly distilled succinaldehyde (25.0 g, 0.290 mol, 1.00 equiv) and EtOAc (390 mL, 0.75 m) (Note 14). The solution is briefly stirred (800 rpm, 30 sec) to ensure full dissolution of succinaldehyde and formation of a homogenous solution (Note 15). To the rapidly stirring solution is added 1,3,5-trimethoxybenzene (1.22 g, 7.25 mmol, 2.50 mol%) as an internal standard, followed by l-Proline (669 mg, 5.81 mmol, 2.00 mol%). The reaction mixture is stirred (800 rpm) at room temperature for 40 h (Figure 4) (Note 16).
Figure 4. Visual appearance of the reaction following stirring at room temperature for 40 h (photo provided by checkers)
The reaction mixture is subsequently diluted to 0.35 m by the addition of EtOAc (440 mL) and an aliquot (0.1 mL) is taken to check for conversion of succinaldehyde (Note 17). Thiomorpholine trifluoroacetate (1.26 g, 5.81 mmol, 2.00 mol%) (Note 18) is then added before the reaction mixture is placed into a pre-heated IKA heating block (70 °C) and stirred for 2 h (Notes 19 and 20) (Figure 5).
Figure 5. A) Reaction appearance after 5 min of heating, B) Reaction appearance after 2 h of heating (photos provided by checkers)
Following this, an aliquot (0.1 mL) is taken to check for conversion of succinaldehyde and the NMR yield of bicyclic enal 2 (Note 21). Pre-treated wet silica gel (Note 22) is then added, and the reaction mixture is removed from the heating block and allowed to cool to room temperature (Note 16) while stirring vigorously for 30 min (Note 23). The crude reaction mixture is filtered through a Büchner funnel into a 2 L filter flask equipped with a 4.0 cm oval magnetic stir bar. The reaction flask and filter cake are then washed thoroughly with EtOAc (2 × 250 mL) (Note 24). Aqueous Na2SO4 (500 mL, 12% w/w) (Note 25) is added to the mother liquor and the biphasic mixture is stirred vigorously for 10 min before transferring to a 2 L separating funnel and allowing the phases to separate (Note 26). The aqueous phase is separated, and the organic phase is collected. Extraction of the aqueous phase is repeated two more times with EtOAc (2 × 500 mL) and the organic phases are combined, dried with MgSO4, filtered, and concentrated under reduced pressure (7.5 mmHg, 25 °C) to afford a crude brown oil (Figure 6).

Figure 6. TLC of the crude reaction mixture (50% EtOAc in n-pentane) stained with p-anisaldehyde showing bicyclic enal 2 alongside succinaldehyde (photo provided by checkers)
The crude product is placed under high vacuum (0.01 mmHg) for 1 h to facilitate further removal of succinaldehyde prior to flash column chromatography (Note 27). Crude bicyclic enal 2 is charged directly onto a column (5 × 45 cm) containing pre-treated wet silica gel (150 g) (Note 22) using dichloromethane (25 mL) to facilitate transfer. The desired product is eluted with 50% EtOAc in n-pentane (45 mL fractions) and obtained in fractions 30-72 (Note 28). The fractions containing product are combined and concentrated under reduced pressure (7.5 mmHg, 25 °C) to afford bicyclic enal 2 (5.76 g, 26% yield, >99:1 e.r., 1.94:1 d.r.) as a light brown solid (Notes 29, 30, and 31) (Figure 7).
Figure 7. Visual appearance of bicyclic enal 2 following flash column chromatography (photo provided by checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
2,5-Dimethoxytetrahydrofuran, deionized
H2O,
toluene,
succinaldehyde, dry ice,
acetone,
ethyl acetate,
1,3,5-trimethoxybenzene,
l-Proline,
Thiomorpholine,
thiomorpholine trifluoroacetate, silica gel,
sodium sulfate,
magnesium acetate,
dichloromethane,
n-pentane,
methanol,
chloroform-d,
potassium carbonate,
acenaphthene,
diethyl ether, and
Trifluoroacetic acid.
2.
2,5-Dimethoxytetrahydrofuran (mixture of
cis- and
trans-isomers, 99%) was purchased from Acros Organics and used as received.
3. All exposed hot surfaces were insulated with cotton wool and aluminum foil to ensure constant distillation.
4. A distillate (mixture of
H2O and
MeOH) of 110 mL was collected during this period.
5. Attempts to remove the remaining solvent by distillation required lower pressures and higher temperatures, which facilitated polymerization and more polymer content in
succinaldehyde. Therefore, rotary evaporation was used for further removal of solvent, which did not interfere with the yield and purity.
6. The reaction mixture was allowed to cool to 65 °C over the course of 45 minutes. The distillation apparatus was disassembled, and the flask was connected to a rotary evaporator with the water bath set to 65 °C. The pressure was reduced in a slow and controlled manner to ensure the reaction mixture did not bump.
7. Caution:
succinaldehyde has a particularly distinct and unpleasant odor. At this stage, crude
succinaldehyde can be diluted with
dichloromethane (ca. 4 mL/g) and stored in a freezer (-20 °C) prior to distillation the following day. The crude
succinaldehyde can be stored in
dichloromethane at -20 °C for up to a month, but it must be distilled, and purity assessed by NMR prior to use in the next reaction. Distillation temperature and vacuum must be carefully maintained to prevent any polymerization
8. Crude
succinaldehyde was transferred to the 100 mL round-bottomed flask using
dichloromethane to facilitate transfer. The
dichloromethane was then carefully removed under reduced pressure (22 mmHg, 25 °C) and the crude product was stirred under high vacuum (0.1 mmHg) for 30 min to ensure thorough removal of
dichloromethane and residual
toluene prior to distillation. Care should be taken during distillation to avoid bumping of the crude product over to the receiving flask. If this is a problem, it can be mitigated by gradual heating or use of a larger distilling flask (e.g. a 250 mL round-bottomed flask).
9. The dry ice/
acetone bath is necessary to prevent potential polymerization of neat
succinaldehyde during the distillation process.
10. All exposed hot surfaces were insulated with cotton wool and aluminum foil to ensure constant distillation. Towards the end of the distillation process, the oil bath can be increased to 90 °C to assist with the complete distillation of
succinaldehyde.
11. Following completion of distillation, the dry ice/
acetone bath was removed and the receiving flask containing the colorless solid
succinaldehyde was allowed to warm under high vacuum to provide
succinaldehyde as a colorless oil. This slow-warming process also prevents the rapid polymerization of neat
succinaldehyde.
12.
Succinaldehyde can be stored as a solution in
dichloromethane (ca. 4 mL/g) and kept in a freezer (−20 °C) for 4 weeks. However, to ensure reliable and reproducible results,
succinaldehyde must always be freshly distilled prior to use and its quality checked by
1H NMR analysis. The quality of distilled
succinaldehyde can be determined by
1H NMR analysis using
13C-
1H satellites in a manner similar to that described by Davies and co-workers.
3 13C-
1H satellites are 1.108% abundant and provide a ratio of 1:178.5
vs. the parent
12C-
1H resonance. Integration of one of the
13C-
1H satellites of
succinaldehyde's aldehydic hydrogen resonance (d = 9.82 ppm,
chloroform-d) to provide a value of 1.00 allows for comparison against the integration of the oligomeric hemi-acetal region (d ≈ 5.80 - 5.30 ppm,
chloroform-d). Where the integration of oligomeric material is ≤15.00, the subsequent
l-Proline-catalyzed dimerization will provide yields between 25-29% as described. Freshly distilled
succinaldehyde always shows the oligomeric proton integration ≤6.00. But storage period of >45 min causes the integration to increase >15.00. For this and subsequent analysis, the
chloroform-d used was treated with
K2CO3 to remove any DCl, which can promote
succinaldehyde oligomerization as well as decomposition of bicyclic enal
2, which is prepared in the next step.
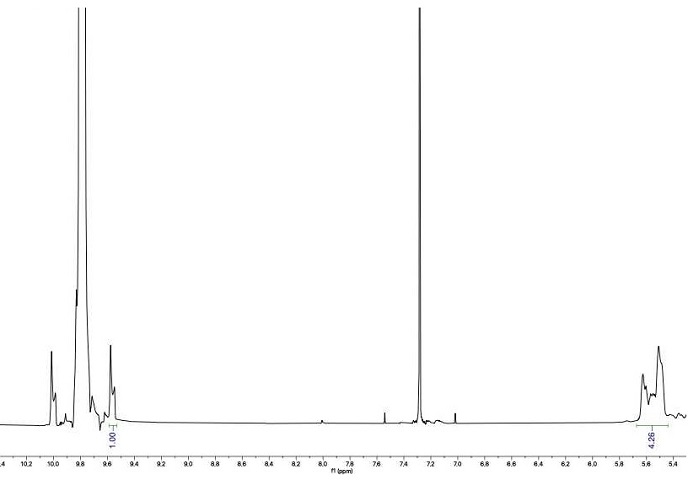
If excess oligomers are observed in the 1H NMR, succinaldehyde is distilled again. In our experience, the re-distillation of succinaldehyde can lead to bumping and as such, a short solvent still head placed between the distilling flask and condenser can mitigate this.
13.
Succinaldehyde has the following physical and spectroscopic properties:
Rf = 0.34 (50%
EtOAc in
n-pentane);
1H NMR
pdf (400 MHz,
chloroform-d) d: 9.80 (s, 2H), 2.79 (s, 4H);
13C NMR
pdf (101 MHz,
chloroform-d) d: 199.8, 36.2; IR (neat)
νmax: 2910, 2839, 2734, 1711, 1387, 1355, 1261, 1195, 1052, 979, 925, 870 and 764 cm
−1. Quantitative
1H NMR
pdf analysis using 14.0 mg of
succinaldehyde and 26.0 mg of
acenaphthene (99%, Sigma Aldrich) as an internal standard provided a purity assessment of 93.3% by weight. Two other reactions performed by the checkers on full scale provided 48.8 g (73%) and 55.9 g (84%) of
succinaldehyde.
14.
Succinaldehyde (25.0 g, 0.290 mol, 1.00 equiv) was transferred to the 2 L round-bottomed flask using a glass Pasteur pipette as previous transfers using a syringe and needle had resulted in the rapid polymerization of neat
succinaldehyde. The quality of
succinaldehyde must be checked by
1H NMR analysis prior to setting up the
l-Proline catalyzed dimerization as the presence of oligomers will result in the observable formation of numerous purple oligomers that stick to the walls of the round-bottomed flask and result in a poor yield of bicyclic enal
2.
15. It is essential to ensure full dissolution of
succinaldehyde in
ethyl acetate prior to the addition of
l-Proline (free-flowing solid) (ReagentPlus®, ≥99%; purchased from Sigma-Aldrich). This can be confirmed by briefly stirring the solution (800 rpm, 30 seconds to 2 min) and observing the dissolution of
succinaldehyde to form a homogenous solution. If this is not performed, rapid formation of pink/purple oligomers is observed.
16. Room temperature was typically (20-24 °C).
17. An aliquot (0.1 mL) of the reaction mixture was taken and concentrated under high vacuum (0.01 mmHg) to remove
ethyl acetate.
chloroform-d was added (0.5 mL) and the solution transferred to an NMR tube.
1H NMR analysis using a Bruker 400 MHz NMR spectrometer (16 scans, 30° pulse angle, 30 s relaxation delay, 25 °C) followed by integration of the aromatic signal of
1,3,5-trimethoxybenzene (d = 6.08, s, 3H), then the aldehyde of
succinaldehyde (d = 9.82, s, 2H) quantified the amount of
succinaldehyde remaining by the following method: The integration of the
succinaldehyde peak was divided by 80 to account for both the 2.50 mol% of internal standard used and the two aldehyde protons of
succinaldehyde. The value obtained was then multiplied by 100 to convert into a percentage. At this point in the reaction, 10% of
succinaldehyde remained.
18. Thiomorpholinium trifluoroacetate was prepared according to a procedure outlined by List and co-workers:
4 To a rapidly stirring solution of freshly distilled
Thiomorpholine (1.00 mL, 10.0 mmol, 1.00 equiv) in anhydrous
diethyl ether (20 mL) was added a solution of
Trifluoroacetic acid (0.84 mL, 11 mmol, 1.10 equiv) in anhydrous
diethyl ether (10 mL) at 0 °C slowly dropwise (syringe pump: 1 mL/min). The reaction was allowed to stir at 0 °C for 1 h before warming to room temperature and stirring overnight. The white precipitate obtained was filtered and washed thoroughly with
diethyl ether before leaving to dry under high vacuum (0.1 mmHg) overnight to afford thiomorpholinium trifluoroacetate (2.12 g, 98%) as a white solid.
19. The reaction temperature reaches ~65 °C after 30 min of being immersed in the oil bath and is stirred for 2 h from this point.
20. The reaction mixture color turns from a luminous red (5 min) to a deep purple (15 min), and finally, to a brown heterogeneous mixture (30 min).
21. An aliquot of the reaction mixture (0.1 mL) was taken at this point to quantify the amount of
succinaldehyde remaining using the method described in
Note 12 (5% of
succinaldehyde remained). The NMR yield of bicyclic enal
2 was also obtained using the same method: following integration of the aromatic signal of
1,3,5-trimethoxybenzene (δ = 6.08, s, 3H), the diastereomeric alkene protons of bicyclic enal
2 (δ = 6.78,
app. q, 1H; δ = 6.65,
app. q, 1H; 1.9:1 d.r.) were integrated and the values obtained added together. This number was divided by 20 to account for the 2.50 mol% of internal standard used and the 0.5 equivalents of bicyclic enal
2 formed (dimerization). The value obtained was then multiplied by 100 to convert into a percentage. At this point in the reaction, a 30% NMR yield of bicyclic enal
2 was obtained.
22. Pre-treated wet silica gel was prepared by adding H
2O (25 mL) to silica gel (50 g; Sigma-Aldrich technical grade, 40-63 µm) (33%
w/w) inside a sealable container. The container lid was closed, and the mixture was shaken vigorously until a uniform consistency was obtained (this process is exothermic, and care should be taken when venting the container). The wet silica gel was then allowed to stand for 1 h prior to use.
23. The pre-treated wet silica gel helps by binding the polymers produced during the dimerization process as well as assisting with the decomposition of
succinaldehyde.
24. An aliquot (0.1 mL) of the mother liquor can be taken at this point and analyzed according to Notes
17 and
21 to quantify the level of
succinaldehyde remaining and the NMR yield of bicyclic enal
2 to ensure thorough removal of the desired product from the wet silica gel.
25. The 12%
w/w aqueous Na
2SO
4 solution was prepared by slowly adding Na
2SO
4 (120 g) portion wise to vigorously stirred H
2O (880 mL). Gentle heating was used to assist with the dissolution of Na
2SO
4. Previously, 17%
w/w aqueous Na
2SO
4 solutions were used for this "salting-out" extraction process.
5,6 However, this increased saturation often led to Na
2SO
4 crashing out of the aqueous phase during extraction.
26. An aliquot (the tip of a glass Pasteur pipette) of the crude product can be taken at this point and analyzed according to Notes
17 and
21 to quantify the level of
succinaldehyde remaining and the NMR yield of bicyclic enal
2.
27. Residual
succinaldehyde co-elutes with the product during flash column chromatography and also causes streaking.
28. Following elution of fraction 30 (~1.5 L of eluent) the gradient can be increased to 75%
EtOAc in
n-pentane to facilitate quicker removal of product. However, this results in bicyclic enal
2 being obtained in slightly lower purity as a dark brown solid.
29. Bicyclic enal
2 has the following physical and spectroscopic properties: mp 88-90 °C;
Rf = 0.20 (50%
EtOAc in
n-pentane);
1H NMR
pdf (400 MHz,
chloroform-d) d: 9.77 (s, 0.3 × 1H),* 9.76 (s, 0.7 × 1H), 6.78 (
app. q,
J = 2.1 Hz, 0.3 × 1H),* 6.65 (
app. q,
J = 2.1 Hz, 0.7 × 1H), 5.56 (d,
J = 5.0 Hz, 0.7 × 1H), 5.52 (dd,
J = 4.8, 0.3 × 1H),* 4.96 (broad t,
J = 6.1, 0.7 × 1H), 4.89 (dt,
J = 6.9, 4.1 Hz, 0.3 × 1H),* 3.66 (dt,
J = 11.0, 6.0 Hz, 0.7 × 1H), 3.59-3.52 (broad m, 0.3 × 1H),* 3.08 - 2.66 (m, 3H),* 2.33 - 2.17 (m, 1H),* 2.10 (d,
J = 13.3 Hz, 0.3 × 1H),* 1.94 (dt,
J = 13.3, 5.1 Hz, 0.7 × 1H) ppm (
peaks which contain minor diastereomer have been marked with an asterisk (*));
13C NMR
pdf (101 MHz,
chloroform-d):
δ 190.2,* 190.1, 153.3,* 152.4, 144.7,* 144.7, 99.0,* 98.6, 82.9,* 80.9, 50.0,* 49.4, 38.2,* 38.0, 35.5 ppm (
peaks which contain minor diastereomer have been marked with an asterisk (*)); HRMS (ESI)
m/z calculated for C
8H
10NaO
3 [M+Na]
+: 177.0522, found: 177.0526; IR,
νmax: 3468, 2993, 2928, 2855, 1659, 1617, 1444, 1367, 1261, 1162, 1086, 1036, 995, 979, 870, 797, 764 cm
-1. Quantitative
1H NMR
pdf analysis using 25.0 mg of bicyclic enal
2 and 25.0 mg of
acenaphthene (99%, Sigma Aldrich) as an internal standard provided a purity assessment of 92.0-94.0% by weight. This can be improved to 98.5 wt.% purity following recrystallization from
toluene.
30. The enantiomeric ratio of the enal was determined following chiral GC analysis as described by Aggarwal and co-workers.
5,7 Chiral GC analysis showed the enantiomeric ratio to be >99:1: Supelco Beta Dex™ 325, Fused silica capillary column (30 m x 0.25 mm x 0.25 µm film thickness); Gas - N
2, constant pressure 20 psi, Inlet temperature: 250 ºC, Split ratio 10:1, Detector: FID 250 ºC, Temperature regime: Start 70 ºC (3 min hold), heating to 200 ºC with speed 3 ºC/min (10 min hold). Sample concentration ~ 0.25mg/mL,
tR = 36.749 (major),
tR = 36.420 (minor). The other isomer was prepared using D-proline as the catalyst following the identical procedure.
31. A second reaction by the checkers on the same scale provided 5.61 g (25%) of the light brown solid.
3. Discussion
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved