The synthesis of C-C bonds by cross-coupling of carbon nucleophiles with carbon electrophiles has revolutionized organic synthesis. While C(sp
2)-C(sp
2) bond formation has been well-developed, C(sp
2)-C(sp
3) bond formation has become increasingly important to drug discovery. While advancements have been made in carbon nucleophile synthesis, it remains the case that carbon electrophiles are more abundant than carbon nucleophiles. An increasingly useful solution is found in the cross-electrophile coupling (XEC) of C(sp
2) and C(sp
3) electrophiles, exemplified by Step B in our original
Org. Synth. publication above. In these XEC reactions, cross-selectivity depends upon the relative ease of radical formation rather than electronic differences of the coupling partners, providing a complementary tool for expanding chemical space. This update provides an overview of the many advancements in the XEC of aryl (and vinyl) (pseudo)halides with alkyl halides (and other alkyl radical equivalents) with an emphasis on practical advancements. As this field is rapidly expanding, more comprehensive treatments of XEC reactions are available for further reading.
2,3,4,5Mechanism
At the time of our original report, the mechanism of XEC of aryl halides with alkyl halides was poorly understood. Studies by our group had shown that these reactions did not proceed via an organozinc intermediate,
6,7 ruling out established Ni-catalyzed cross-coupling mechanisms. Informed by key prior electrochemical studies,
8,9 further studies suggested a new type of mechanism (Figure 1).
10,11 Subsequent studies
2,12,13 have largely converged on three essential elements:
1) The alkyl electrophile is converted into an alkyl radical intermediate, and the aryl or vinyl electrophile reacts with the Ni catalyst to form an (L)Ni
II(Ar)X intermediate by a non-radical mechanism.
2) The alkyl radical reacts with the (L)Ni
II(Ar)X intermediate to form a putative (L)Ni
III(Ar)(Alkyl)X intermediate that rapidly undergoes reductive elimination to form the C(sp
2)-C(sp
3) bond.
3) The Ni catalyst is reduced to turn over the catalytic cycle.
Figure 1. General proposed mechanism for C(sp2)-C(sp3) XEC
While the exact details of this mechanism are still a source of active investigation and debate, its study has led to improved understanding of the mechanistic steps involved.
3,11,13,14 This understanding has led to expansion of the substrate classes that participate in the transformation, enantioselective methods, an explosion of related variants such as metallaphotoredox and electrochemical approaches, and applications in total synthesis.
New Radical Precursors
While alkyl halides are far more abundant than alkyl organometallic reagents, the largest commercially available pools of aliphatic electrophile diversity are alcohols, amines, and alkanoic acids. A major, recent advance has been the development of methods to cross-couple these substrate pools, sometimes with a simple in situ activation step (Figure 2).
Figure 2. Radical precursors used in XEC coupling reactions
While conversion of alcohols to alkyl halides is often high yielding, in parallel synthesis applications the extra steps and purifications are limiting; on scale, the extra time and solvent is a large cost. Directly utilizing alcohols without stoichiometric activation has been limited to more activated substrates, such as allylic and benzylic alcohols. Ukaji reported the coupling of benzylic alcohols with aryl iodides enabled by a low-valent Ti Lewis acid.
15 Both Shu and Wang reported a similar approach for the reaction between allylic alcohols and aryl halides, instead employing Zr and Mg Lewis acids, respectively.
16,17 For reactions with unactivated alcohols, many groups have focused on the coupling of redox-active alcohol derivatives or in situ generation of alkyl halides.
8,18,19 Martin reported the use of
N-alkoxyphthalimides which, upon reduction, generate
O-centered radicals that undergo
β-scission to generate alkyl radicals that engage in XEC.
20 Gong utilized dialkyl oxalates which can undergo Barton C-O scission to generate 3° and stabilized alkyl radicals capable of engaging in XEC with aryl halides.
21,22 Molander reported the Ni-catalyzed XEC of unactivated alkyl tosylates with aryl and heteroaryl bromides using KI as a mediator, generating the alkyl iodide in situ.
23 We reported a protocol for the coupling of benzyl mesylates with halides using co-catalytic cobalt phthalocyanine (Co(Pc)).
24 The use of tosylates has been used in several industrial applications since these original reports.
25,26 The in situ conversion of alcohols to alkyl bromides has been reported using three different strategies: 1) the Li group used a paired electrolysis strategy to generate Ph
3PBr
2 in situ for concurrent bromination and XEC, avoiding the use of a metal terminal reductant,
27 2) we used a phosphonium reagent and Bu
4NBr for a rapid in situ pre-bromination step followed by XEC, which was amenable to high-throughput experimentation (HTE) in the solution phase,
28 and 3) Gong and Ma utilized a oxazolium bromide for a fast pre-bromination step, which proved useful for the selective monofunctionalization of diols.
29Alkyl amines have also proven to be a ready source of alkyl radicals through the reduction of the corresponding pyridinium salt. These salts, initially described by Katritzky,
30 are readily prepared through condensation with commercially available pyrylium salts and have been shown to furnish alkyl radicals following single electron reduction.
31,32 Watson and co-workers first described the use of these pyridinium salts in their Ni-catalyzed cross-coupling with aryl boronic acids, demonstrating radical formation with Ni in a redox-neutral coupling.
33 Following this report, several groups concurrently reported Ni-catalyzed XEC methods of aryl iodides and bromides with alkyl pyridinium salts.
32,34,35,36,37 Martin's work suggests that neither on-cycle Ni
0 or Ni
II intermediates are capable of reducing pyridinium salts to form alkyl radicals. Rueping shows theoretical support for the proposed mechanism that these methods share, while the work by Han displays an expanded scope to include alkyl halides and alkynyl bromides as coupling partners. Molander utilized an organic photocatalyst and Et
3N as the terminal reductant in combination with Ni to induce the reaction without a metal reductant. The use of alkyl pyridinium salts has also been extended to the coupling of acyl electrophiles to synthesize functionalized ketones.
38,39Based upon seminal studies in photoredox catalysis by Okada and Overman,
40,41 we reported the use of
N-hydroxyphthalimide (NHP) esters in XEC reactions with aryl iodides.
42 These NHP esters can be readily prepared via condensation with
N-hydroxyphthalimide. Following single electron reduction, these esters fragment to release CO
2, phthalimide anion, and an alkyl radical. NHP esters react with Ni catalysts at rates similar to alkyl iodides, and we reported that reactivity can be modulated by introducing electronic-tuning groups on the phthalimide backbone and by adjusting the solvent polarity.
43 These esters have been coupled to alkynyl bromides
44 and acyl electrophiles, such as anhydrides and 2-pyridyl thioesters.
45,46We demonstrated that a 2° alkyl radical, formed via epoxide opening using a titanocene co-catalyst, can be coupled with (L)Ni
II(Ar)Br species to form a new C-C bond.
47 The opening of epoxides and aziridines can also be achieved through the use of an iodide co-catalyst, which provides an iodohydrin or
β-iodoamine, respectively, that can participate in the XEC as previously described.
47,48 Additionally, redox active sulfones have been shown to generate alkyl radicals following single electron reduction, which can then engage in XEC with aryl bromides.
49New Catalyst Systems and Conditions
Initial studies on XEC established a set of conditions: amide solvents, bipyridine (bpy) and phenanthroline (phen) ligands, Zn or Mn reductant, and iodide salt additives. Several challenging substrate classes have driven the discovery of new catalysts and conditions that are broadly useful.
Figure 3 contains ligands that have been reported for the coupling aryl halides with alkyl radical precursors. Pyridyl-2-carboxamidine (PyCam) and pyridyl-2,6-bis(carboxamidine) (PyBCam) ligands, found via a screen of Pfizer's compound collection for new nitrogen ligand motifs,
50 are useful for aryl halides with coordinating groups in the 2-position and for a variety of heteroarenes.
51 BpyCam (2,2'-bipyridine-6-carboxamidine) derivatives and
N-cyano carboxamidine ligands have also shown promise in a growing collection of challenging reactions.
52,28 Dual ligand strategies commonly include combinations of bidentate
N-donor ligands with phosphines,
6,53 terpyridines,
54 and pyridines.
23,55 For the cross-coupling of unstrained 3° alkyl halides, substituted pyridines are among the best available ligands.
55,56 Finally, Sevov reported on the use of a Ni "overcharge protector" complex, which prevents over-reduction of the Ni catalyst.
57Several groups have reported on conditions where the alkyl radical is formed by a co-catalyst or stoichiometric additive, independent of the Ni catalyst. In several cases, pyridinium salts and NHP esters can be directly reduced by Mn or Zn to form alkyl radicals.
32,58 We demonstrated that benzyl mesylates, which are unreactive with (bpy)Ni catalyst, could be activated by Co(Pc) co-catalysis to furnish diarylmethanes.
24 Co(Pc) is capable of undergoing nucleophilic substitution with benzyl mesylates, after which homolysis of the Co-C bond furnishes the benzyl radical. This radical can then be captured by the (bpy)Ni
II(Ar)Br species and form product upon reductive elimination. Komeyama extended this work to unactivated alkyl tosylates by using the more nucleophilic vitamin B
12.
59 Hazari and Zultanski further demonstrated the utility of Co(Pc) in the organoreductant-promoted coupling of unactivated alkyl bromides with aryl bromides.
60 The Molander group has shown NHP esters can form electron-donor-acceptor complexes with Hantzsch ester which, upon photoexcitation, can generate alkyl radicals that then interact with the Ni catalyst for productive cross-electrophile coupling.
61 Our group and others have used Ni/Ti co-catalysis to generate alkyl radicals through the ring-opening of epoxides and demonstrating their coupling with aryl halides.
62,63 Ni/photoredox co-catalysis has emerged as a powerful tool to form C-C bonds centered around decoupling radical and polar elementary steps. Lei, Vanucci, and Molander have reported metallaphotoredox XEC reactions using aliphatic amines as the terminal reductant.
37,64,65 MacMillan reported on a Ni/Ir catalyst system enabled by the use of silane reagents to facilitate alkyl radical formation from alkyl halides by halogen-atom transfer to a silicon-centered radical.
66,67 By tuning the silane, this approach has been extended to other substrates.
68,69
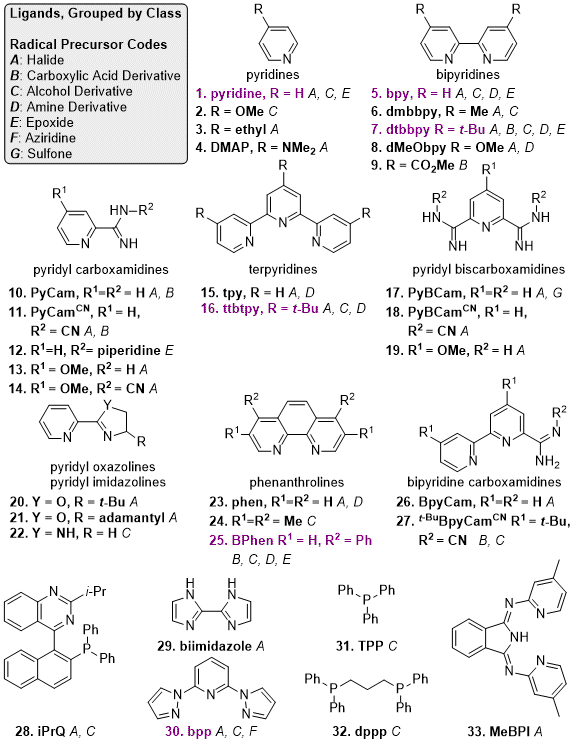
Figure 3. Reported ligands for C(sp2)-C(sp3) XEC. Ligands that have been used with at least three different radical precursors are highlighted in purple
New Reductants
The most common electron sources used in XEC for catalyst turnover are metal reductants, typically Zn or Mn powder, which are inexpensive, easy to handle and store on the benchtop, possess good atom economy (27-33 g per mole of electrons), and the resulting metal salts are easily separated from products. However, in >100 g scale applications, these heterogenous reductants present mass-transfer limitations, sometimes resulting in irreproducible kinetics,
70,71,72 and disposal of large amounts of metal salts can be complicated.
73,74 In addition, accurately characterizing the activity of metal powders is difficult, leading to lot-to-lot variability.
The use of organic terminal reductants has been explored, resulting in several different systems reported recently, such as pure organic reductants, photoredox-assisted organic reductants, and electrochemically driven organic reductants (Figure 4). These alternative reduction systems often allow for non-amide solvents to be used, suggesting solvent limitations are tied to the reductant rather than the catalyst. We utilized tetrakis(dimethylamino)ethylene (TDAE) in mechanistic studies to support that an organozinc intermediate was not necessary for XEC.
10 We also found that the use of TDAE as reductant in XEC allows for the use solvents such as acetonitrile and propylene carbonate.
73 Reisman demonstrated that TDAE could drive XEC reactions between NHP esters and alkenyl bromides.
75 Recent work by Hazari, Uehling, Zultanski and coworkers reported the synthesis and use of tuned homogeneous reductants based on the tetraaminoethylene scaffold seen in TDAE.
60 These new reductants exhibit a range of reduction potentials as well as increased air-stability, allowing for use and storage on the benchtop. A variety of groups have reported on the use of sacrificial Zn, Fe, or steel anodes for XEC reactions.
76 While this avoids issues with stirring metal powders, it still results in stoichiometric metal salt waste. Some recent reports have utilized an amine terminal reductant in a divided electrochemical cell.
54,77 Several groups concurrently reported on the use of photoredox catalysis to allow the use of organic terminal reductants, such as silanes and 3° amines.
37,64,65,66
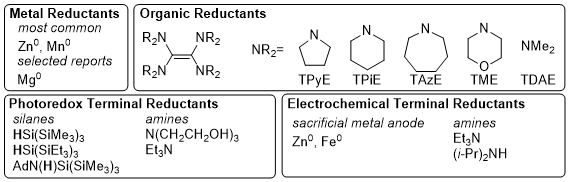
Figure 4. Reductants used in XEC
Enantioconvergent, Enantioselective, and Enantiospecific Reactions
XEC reactions that allow setting a stereocenter have progressed rapidly alongside achiral work, often providing useful mechanistic insights as well as powerful new transformations. Although there exist parallels to enantioconvergent cross-coupling reactions with nucleophiles, realization of enantioconvergent XEC of racemic 2° electrophiles proved to be complex. Initial studies by the Reisman group demonstrated the key concepts that set the stage for the present blossoming of the field. In particular, 2° benzylic radical precursors have been the most reliable C(sp
3) coupling partners for a variety of C(sp
2) coupling partners.
78,79,80,81,82 Coupling of aryl halides with α-halo nitriles,
83 α-halo esters,
84 and α-halo sulfones
85 also appear quite general. In a twist on benzylic radical precursors, the stereoconvergent XEC of styrenyl aziridines with aryl iodides using a chiral BiOx ligand was also reported.
48 Recent work has included biimidazoline as a competent chiral ligand for the XEC of aryl or alkenyl halides with alkyl chlorides or styrene oxides under metallaphotoredox conditions.
86,87 Several chiral ligand classes have allowed for enantioconvergent reactions (Figure 5).
88,89Beyond enantioconvergent XEC, there are a few reports of enantioselective XEC. We have reported the enantioselective XEC of
meso-epoxides with aryl bromides via a chiral Ti co-catalyst.
62 Based upon studies by Gansauer,
90 we proposed that this co-catalyst engaged in the enantioselective opening of the
meso-epoxide, forming a
β-titanoxy carbon radical, followed by radical capture by the (L)Ni
II(Ar)Br complex and reductive elimination to generate the
trans-
β-arylcycloalkanol.
Figure 5. Chiral ligands utilized in enantioconvergent XEC
In 2016, the Jarvo lab reported the first enantiospecific XEC reaction (the intramolecular coupling of benzylic esters with aryl bromides) which proceeds via inversion at the benzylic center.
91 Despite many advancements in enantiospecific, intramolecular C(sp
3)-C(sp
3) coupling, enantiospecific XEC of aryl and alkyl halides remains limited thus far.
5,92Adaptation to Small and Large Scale
Interest in applications to medicinal and process chemistry has driven innovation in reaction solvent, reactor design, and reductant choice. The outcomes of these approaches have demonstrated areas for improvement.
Medicinal chemistry routinely uses parallel, small scale (≤~10 µmol) reactions to rapidly explore chemical space and process chemistry often uses the same parallel systems to optimize a key step. Adapting XEC to HTE revealed difficulties in stirring heterogeneous reactions, dosing small amounts of metal powders, and identifying additional catalysts to cover larger areas of chemical space.
93 A number of reports addressed these issues by slurrying the metal powder and dosing the slurry with a large-bore pipette, which is sufficient as long as excess reductant is employed, and others utilized stirring with tumble stirrers.
93 Another solution is coating the reductant onto ChemBeads
94 (small glass beads) and then dosing by weight or volume (with a scoop); these have the added advantage of allowing the use of a shaker instead of a tumble-stirrer (Figure 6).
9395 This approach has proven to be general for several different XEC reactions and is relatively affordable to implement.
28,43,96
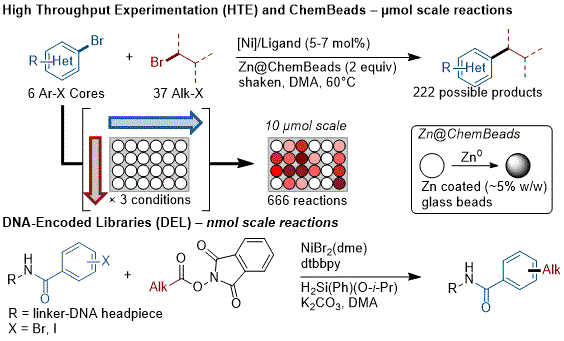
Figure 6. Strategies for small-scale XEC at µmol and nmol scale
On the very smallest of scales, DNA-encoded libraries (DELs) have rapidly become a key tool in medicinal chemistry, but present unique challenges for reaction development as they usually require aqueous conditions, high salt concentrations, and very low substrate (DNA) concentrations.
97 This has been addressed by utilizing reversible adsorption to solid support (RASS) with a silane terminal reductant, allowing the use of amide solvents without water or photochemical activation (Figure 6).
98 The other main approach has been the use of metallaphotoredox catalysis (vide supra).
99,100 Overall, XEC reactions appear amenable to DELs and we expect to see additional reports in this area in the future.
On larger scales, researchers have explored both batch and flow processes. Flow chemistry has become a key tool in the pharmaceutical industry for scale up and XEC has been adapted in several different ways, dependent upon the reduction approach: a packed Zn column,
101 a flow electrochemical cell
77 or a flow photochemical cell.
102,103,104 Thus far, these flow reactions have been largely proof of concept and not yet scaled further, suggesting a promising area for future development.
Following our original report in
Org. Synth., reports on batch scale up reactions have also appeared. Most have used metal reductants (Mn or Zn), but sacrificial anode electrochemical reactions have also been explored.
25,76 With metal powders, specialized stirring equipment and metal powder activation have been important to success as the reactor size (and shape) changes.
25,70 To date, the largest reported XEC reaction is a batch reaction to produce 5.7 kg of product (64% yield). The reaction used Mn powder as the reductant (activated with TES-Cl) in a 600 L reactor.
70Conclusions
Nickel-catalyzed cross-electrophile coupling has seen significant growth in the past decade. Since the initial report, the substrate scope has expanded to include epoxides, aziridines, and derivatives of carboxylic acids and alkyl amines as alkyl radical sources for coupling. Various modes of activation have been developed, and a clearer mechanistic understanding is being used to drive these advancements. As we and others continue to examine and address the limitations of these methods, we anticipate that cross-electrophile coupling will continue to evolve and become a mainstay in organic synthesis.
That leaves an important question: whither, nucleophiles?
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved