N-Boc-
N-Hydroxymethyl α-amino aldehyde
3 is a configurationally stable α-amino aldehyde,
2 which was successfully converted to
trans-oxazolidine methyl ester
4, a chiral synthon for β-amino-α-hydroxy acids.
3 The
N-hydroxymethyl group on
3 shifts the equilibration to the five-membered cyclic hemiacetal
2, which ensures the high stability of the corresponding α-amino aldehyde and enables an intramolecular conjugate addition for highly diastereoselective introduction of an α-hydroxyl group. As a result,
trans-oxazolidine methyl ester
4 was obtained from
2 as a chiral synthon for
syn-β-amino-α-hydroxy acids.
8,9 Even Garner's aldehyde,
10 one of the most cited stable α-amino aldehydes, not only suffers from 3~5% racemization during the preparation and storage,
3 Other γ-amino-β-hydroxy acids, β-deoxy-β,γ-diamino acid, and α-amino-β-hydroxy acid have been synthesized from
N-Boc-
N-hydroxymethyl α-amino aldehydes.
2,4,5,6 In addition, phenylsulfonylnitromethane (
5) has been used for a two-way homologation of aliphatic aldehydes, which was applied to synthesize precursors for histone deacetylase (HDAC) inhibitors.
7
Configurationally Stable α-Amino Aldehyde
The preparation of configurationally stable α-amino aldehyde for the applications as chiral synthons or auxiliaries has been an important topic in asymmetric synthesis because of the rather acidic α-proton adjacent to the carbonyl group.
8,9 Even Garner's aldehyde,
10 one of the most cited stable α-amino aldehydes, not only suffers from 3~5% racemization during the preparation and storage,
8 but it is also only applicable to certain α-amino acids containing a hydroxyl group such as serine.
We have proposed
N-hydroxymethyl α-amino aldehydes
7 as a suitable candidate for the configurationally stable α-amino aldehyde and undertaken investigations to demonstrate their diverse applications (Scheme 1). Various
N-protected amino acids (
i.e., phenylalanine, valine, serine, leucine and isoleucine) were successfully converted to the corresponding
N-hydroxymethyl α-amino aldehydes
7, whose
N-protecting group could be varied with a Boc, Cbz, or Ts group. As expected,
N-protected
N-hydroxymethyl α-amino aldehydes
7 showed remarkable configurational stability due to the shift of equilibrium to the five-membered cyclic hemiacetal
6 (Scheme 1a).
2,11 For example, phenylalaninal
2 showed 1~1.5% racemization during the preparation, and no racemization during the storage at room temperature for a month.
N-Boc-
N-hydroxymethyl serinal (
7, R
1 = CH
2OSiR
3), an alternative for Garner's aldehyde, also showed less than 1% racemization during the preparation, and no racemization during storage at room temperature for 15 days. In addition, the aldehydes successfully reacted with various nucleophiles via the aldol reaction,
11,12 the nitroaldol reaction,
3,7 and the Wittig reaction.
4,5,6,13,14,15,16γ-Amino-β-hydroxy Acids from N-Hydroxymethyl α-Amino Aldehydes
One of the most useful applications of N-hydroxymethyl α-amino aldehydes 7 was the stereoselective synthesis of a series of γ-amino-β-hydroxy acids 10 (Scheme 1a). The intramolecular conjugate addition of the N-hydroxymethyl group, which was introduced to enhance the stability of α-amino aldehyde, onto γ-amino-α,β-unsaturated esters 8 provided trans-oxazolidines 9 with high diastereoselectivity, presumably through a favored H-eclipsed conformation due to the allylic strain (Scheme 1b).
Scheme 1. Intramolecular conjugate addition with N-Boc-N-hydroxymethyl α-amino aldehydes
This synthetic strategy was first applied to the synthesis of (-)-statine, a key component of a natural antibiotic (Scheme 2a).
4 The intramolecular conjugate addition onto (
E)-α,β-unsaturated methyl ester
12, prepared from
N-Boc-
N-hydroxymethyl-L-leucinal via a Wittig reaction with methyl(triphenylphosphoranylidene)acetate, afforded
trans-oxazolidine
13 (up to
trans:
cis = 10:1). The change in R
1 group in Scheme 1 from a bulky isobutyl group (R
1=-CH
2CH(CH
3)
2, Scheme 2a) to a smaller group (R
1=CH
2OTBS, Scheme 2b) resulted in a decreased diastereoselectivity of
trans-oxazolidine (
trans:
cis = 5:1).
5 However, the reaction onto (
Z)-α,β-unsaturated methyl ester
1517 afforded much higher diastereoselectivity of
trans-oxazolidine
16 (
trans:
cis > 20:1), which was converted to a glutamate receptor modulator
threo-β-hydroxyglutamic acid (Scheme 2b).
5Scheme 2. Synthesis of γ-amino-β-hydroxy acids
Other internal nucleophiles such as an
N-aminomethyl group or
N-hydroperoxymethyl group could be introduced by simple modification of the
N-hydroxymethyl group in γ-amino-α,β-unsaturated ester (Scheme 3).
6,14,15,16 The intramolecular conjugate addition with the
N-aminomethyl group enabled the diastereoselective synthesis of a 3-aminodeoxystatine derivative
20. The intramolecular nucleophilic epoxidation of γ-amino-α,β-unsaturated ester
22 afforded
anti-epoxide
23 with more than 20:1 diastereoselectivity, which was converted to
trans-oxazolidinone
24, a precursor for a glutamate receptor modulator 3,4-dihydroxyglutamic acid, via 5-
exo cyclization with the
N-Boc group.
15,18 Interestingly, a complementary
anti-isomer of γ-amino-β-hydroxy acid could be obtained by the modification of synthetic procedures. The regioselective reduction by Pd-catalyzed hydrogenation of
anti-epoxide
27 gave
anti-γ-amino-β-hydroxy acid (Scheme 3c),
16 whereas the intramolecular conjugate addition of the
N-hyroxymethyl group onto γ-amino-α,β-unsaturated esters gave
syn-γ-amino-β-hydroxy acid (Scheme 2).
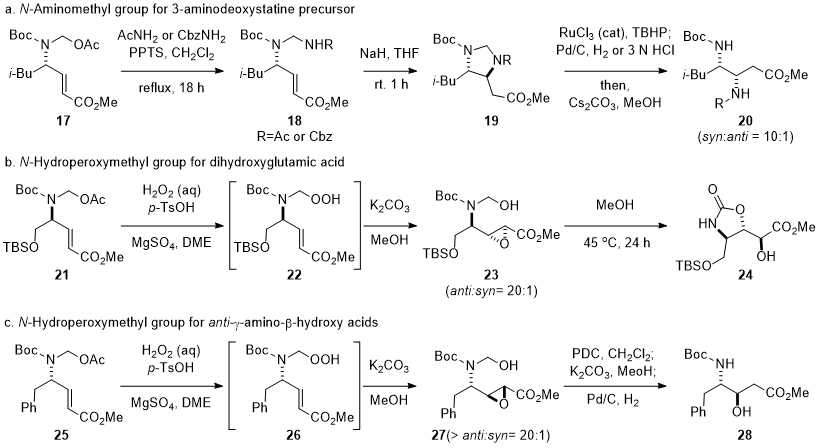
Scheme 3. Modification of N-hydroxymethyl for γ-amino-β-hydroxy acid analogs
β-Amino-α-hydroxy Acids from N-Hydroxymethyl α-Amino Aldehydes
Next, the synthesis of one-carbon lower homologs of γ-amino-β-hydroxy acids, β-amino-α-hydroxy acids, from
N-Boc-
N-hydroxymethyl α-amino aldehydes
29 was explored (Scheme 4a).
3 While γ-amino-α,β-unsaturated esters were used as a Michael acceptor for the synthesis of γ-amino-β-hydroxy acids, nitroolefin derivatives such as
34 prepared with activated nitromethanes (
i.e., PhSO
2CH
2NO
2 (
5)) was chosen as a one-carbon lower Michael acceptor for the synthesis of β-amino-α-hydroxy acids because
N-Boc-
trans-oxazolidine methyl esters
31 could be conveniently obtained by sequential nitroaldol, dehydration, and stereoselective intramolecular conjugate addition reactions under mild basic conditions (
29→
33→
34→
30 in Scheme 4b), followed by one-pot ozonolysis. These sequential one-pot reactions could afford chiral synthons
trans-oxazolidne methyl esters
31 in 65~79% yields with more than 20:1 diastereoselectivity from the corresponding
N-Boc-
N-hydroxymethyl α-amino aldehydes
29, which were readily prepared from α-amino acids (
i.e., phenylalanine, leucine, valine, alanine and serine).
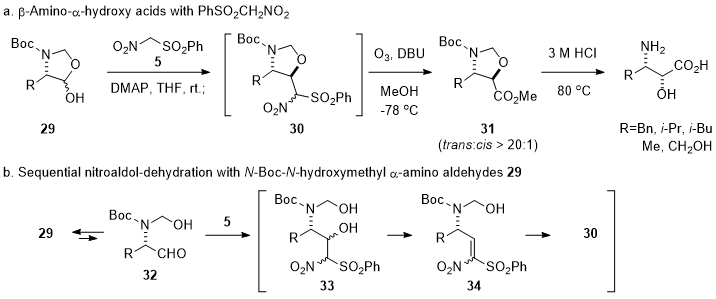
Scheme 4. Synthesis of β-amino-α-hydroxy acids
N-Boc-
trans-Oxazolidine methyl esters
35, a properly protected form of β-amino-α-hydroxy acids, were useful synthons to synthesize derivatives of β-amino-α-hydroxy acid. Basic hydrolysis of
35 followed by the peptide coupling with L-Leu-OMe or L-Val-OMe produced anti-leukemia dipeptide bestatin and its analogs without additional amino- or hydroxy-protecting steps (Scheme 5a).
3,19 Furthermore,
trans-oxazolidine methyl ester
37 derived from D-serine was efficiently converted to L-
threo-hydroxyaspartic acid and L-
threo-hydroxyasparagine after an oxidation of the hydroxymethyl group from deprotection of
37 to the carboxylic acid group of
38 (Scheme 5b).
20 In addition, the orthogonal protecting groups on chiral intermediate
38 made the chemoselective peptide bond formation possible either at the α-amino group (
38→
39→
40), β-hydroxyl group (
38→
39→
41), α-carboxylic acid group (
38→
42), or β-carboxylic acid group (Scheme 5c).
20 Therefore,
trans-oxazolidine dicarboxylate
38 would be useful for the synthesis of bioactive peptides containing an L-
threo-β-hydroxy aspartate moiety, such as WAP-8294A
2, ramoplanin, dolastatins, kahalalide F and neurotoxin antillatoxin.
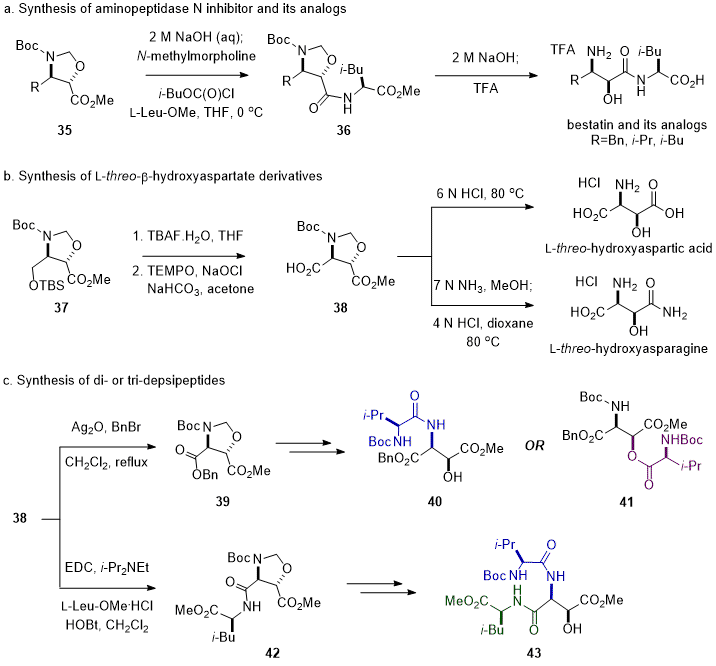
Scheme 5. Application of trans-oxazolidine methyl esters
PhSO2CH2NO2 (5) for One-carbon Homologation
Based on the reaction results between
N-Boc-
N-hydroxymethyl α-amino aldehyde
2 and phenylsulfonylnitromethane (PhSO
2CH
2NO
2 (
5)) as a one-carbon carboxylate synthon, we investigated a new one-carbon homologation of aliphatic aldehydes using PhSO
2CH
2NO
2 (
5). Unlike its reaction with
N-Boc-
N-hydroxymethyl α-amino aldehyde
2,
3,19 the reaction of
5 with aliphatic aldehydes was not successful under similar basic conditions. After enormous attempts, aliphatic aldehydes were successfully converted to β,γ-unsaturated α-nitrosulfone
46 under proline-catalyzed conditions (Scheme 6a).
7 The nitroaldol reactions of
5 with aliphatic aldehydes followed by a dehydration reaction of the nitroaldol intermediate initially yielded α,β-unsaturated α-nitrosulfone
45, which was isomerized to favored β,γ-unsaturated α-nitrosulfone
4621 in the absence of an internal nucleophile. The reaction time was dramatically shortened to 4 h under ultrasound irradiation conditions from 2~3 days with stirring at room temperature.
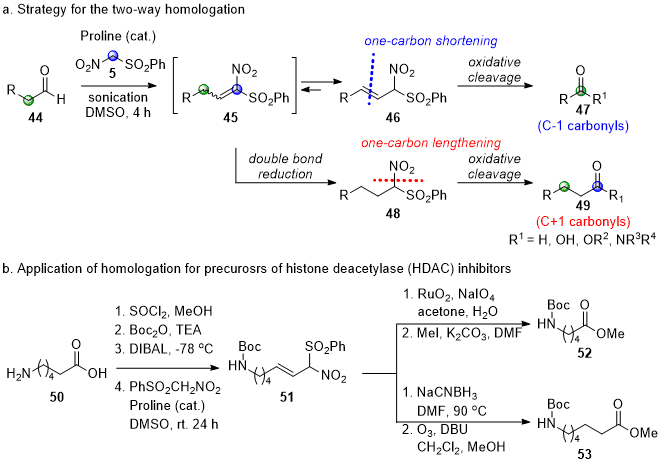
Scheme 6. Applications of PhSO2CH2NO2 (5) for one-carbon homologation
The preference of β,γ-unsaturated α-nitrosulfone 46 over α,β-unsaturated α-nitrosulfone 45 enabled a novel approach toward a two-way homologation of aliphatic aldehydes from the same intermediate β,γ-unsaturated α-nitrosulfone. First, carbonyl compounds (47) with one fewer carbon in the chain were obtained by the ozonolysis of β,γ-unsaturated α-nitrosulfone 46. Four different one-carbon lower carbonyl homologs, dimethyl acetal, aldehyde, carboxylic acid, or primary alcohol, were obtained by quenching ozonides with trimethyl orthoformate and dimethyl sulfide, dimethyl sulfide, hydrogen peroxide, or sodium borohydride, respectively. Next, one-carbon higher carbonyl homologs 49 were obtained by the chemoselective double bond reduction of β,γ-unsaturated α-nitrosulfone 46 followed by the oxidation of phenylsulfonylnitromethyl group to the carbonyl group. The selective reduction was smoothly conducted probably by a conjugate addition of sodium cyanoborohydride onto α,β-unsaturated α-nitrosulfone 45, which was generated after in-situ isomerization of β,γ-unsaturated α-nitrosulfone 46 in DMF at 90 °C (46→45→48 in Scheme 6a). The divergent homologation developed was further applied for the synthesis of the two precursors for biologically active histone deacetylase inhibitors from the common C6-amino acid 50, a C5-amino acid derivative 52 and a C7-amino acid derivative 53 (Scheme 6b).
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved