Org. Synth. 2022, 99, 305-325
DOI: 10.15227/orgsyn.099.0305
Synthesis of Carboxylic Acids from Benzamide Precursors Using Nickel Catalysis
Submitted by Ana S. Bulger, Dominick C. Witkowski, and Neil K. Garg*
1Checked by Cyrus K. Chan, Yu Chen, and Pauline Chiu
1. Procedure (Note 1)
A. N-Methyl-N-phenyl benzamide (3). A single-necked (24/40 joint) 500 mL round-bottomed flask is equipped with a Teflon-coated magnetic stir bar (3 cm, oval-shaped). The flask is dried in an oven (105 °C) for 16 h. The flask is brought out of the oven, sealed immediately with a rubber septum, and allowed to cool to room temperature (23 °C) in a desiccator (Note 2). The flask is purged with argon by connecting to an argon manifold using a needle (18G), and via another needle (18G) to a gas outlet terminating in an oil bubbler. The atmosphere is maintained under a slight positive pressure of argon for the duration of the reaction (Note 3). The flask is then charged with triethylamine (6.5 mL, 47 mmol, 1.3 equiv) (Note 4) using a plastic syringe fitted with a 21G needle, N-methylaniline (2) (4.2 g, 4.2 mL, 39 mmol, 1.1 equiv) (Note 5) via a plastic syringe fitted with a 21G needle in one portion, and dichloromethane (72 mL, 0.50 M) (Note 6) via a glass syringe fitted with an 18G needle. The reaction flask is then placed in an ice bath maintained at 0 °C, and the reaction mixture is stirred for 10 min (250 rpm). Benzoyl chloride (1) (5.1 g, 36.33 mmol, 1.0 equiv) (Note 7) is then added dropwise via a plastic syringe fitted with a 21G needle over 15 min. After 10 min of stirring (250 rpm), the reaction flask is removed from the ice bath, allowed to warm to 23 °C, and the reaction mixture is stirred for 16 h (250 rpm).

Figure 1. Step A reaction setup A) Before the addition of benzoyl chloride, B) 0 min after addition of benzoyl chloride, C) 16 h after addition of benzoyl chloride (photo provided by submitters)
After stirring for 16 h (250 rpm) at 23 °C (Note 8), the reaction mixture is diluted with dichloromethane (200 mL) and 1.0 M HCl (100 mL) (Note 9). The resulting mixture is transferred to a 1 L separatory funnel (24/40 joint) and the layers are separated. The organic layer is washed with a saturated aqueous NaCl solution (2 x 100 mL), dried over Na2SO4 (81 g) (Note 10), and filtered through a fritted Büchner funnel (Note 11) into a 1 L round-bottomed collection flask (24/40 joint). The filtrate is concentrated under reduced pressure on a rotary evaporator to produce 11.2 g of the crude product as an oil (Note 12). Silica gel (14.3 g) is added to the crude oil, followed by the addition of dichloromethane (30 mL). The resulting suspension is concentrated under reduced pressure (Note 13) then placed under high vacuum at 23 °C for 1 h prior to column chromatography. The mixture is then purified via flash column chromatography using hexanes: ethyl acetate as the eluent (Note 14). The eluate containing the product (Note 15) is concentrated under reduced pressure on a rotary evaporator (Note 16). The product is then stored at -20 °C for 14 h, leading to crystallization of the product. The resulting solid is crushed using a spatula and placed under high vacuum for 2 h at 15-min intervals (Note 17) to afford N-methyl-N-phenyl benzamide (3) as a white solid (7.33 g, 96% yield) (Figure 2) (Notes 18, 19, and 20).

Figure 2. Isolated amide 3 (photo provided by submitters)
B. Benzoic acid (5). A single-necked (14/20 joint) 250 mL round-bottomed reaction flask equipped with a Teflon-coated magnetic stir bar (2 cm, oval-shaped), a 5 mL (14/20 joint) round-bottomed flask, and a jacketed reflux condenser (14/20 joint) are dried in an oven (105 °C) for 16 h. The 250 mL flask is capped with a septum and allowed to cool under a stream of dry argon. The jacketed reflux condenser is stoppered with the 5 mL round-bottomed flask, secured with a Keck clip, capped with a septum, and allowed to cool to room temperature under a stream of argon (Note 21). Both sets of glassware are brought into the glovebox (Note 22).
In the glovebox, the 250 mL reaction flask is charged sequentially with N-methyl-N-phenyl benzamide (3) (7.4 g, 35 mmol, 1.0 equiv), 1,3-bis(2,6-di-i-propylphenyl)imidazolidin-2-ylidene (SIPr) (1.4 g, 3.6 mmol, 0.1 equiv) (Note 23) and Ni(cod)2 (1.07 g, 3.90 mmol, 0.11 equiv) (Note 24). The condenser is disconnected from the 5 mL protection flask, and fitted to the 250 mL reaction flask, which is secured using a Keck clip, and the assembled glassware is transferred out of the glovebox (Note 25) into a fume hood. The septum at the top of the reflux condenser is pierced with a needle (18G) connected to an argon manifold, and with another needle (18G) that is connected to a gas outlet terminating in an oil bubbler. The atmosphere is maintained under a slight positive pressure of argon for the duration of the reaction. Under a positive pressure of argon, the Keck clip is removed, and the joint between the flask and condenser is sealed with Teflon tape, then the Keck clip is mounted again. Toluene (35 mL, 1.0 M) (Note 26) is added to the reaction vessel via a plastic syringe fitted with an 18G needle. Finally, 2-(trimethylsilyl)ethan-1-ol (4) (6.2 mL, 43 mmol, 1.2 equiv) (Note 27) is added via a plastic syringe fitted with a 21G needle (Note 28). The condenser is attached to a running water line, and the reaction flask is then placed in an oil bath maintained at 110 °C and stirred for 24 h (250 rpm).

Figure 3. Step B, part 1 reaction setup (photo provided by submitters)
After 24 h, the reaction flask is removed from the oil bath and allowed to cool to 23 °C (Note 29). The reflux condenser is removed from the flask and replaced with a 100 mL (14/24 joint) pressure equalizing addition funnel ((Note 30), Figure 4) under an atmosphere of argon. The addition funnel was charged with TBAF (100 mL, 1.0 M in THF, 2.8 equiv) (Note 31) via a plastic syringe fitted with a 18G needle. The TBAF solution is then added dropwise through the addition funnel to the stirring solution (250 rpm) over 25 min at 23 °C.
Figure 4. Step B, part ii reaction setup A) before addition of TBAF and B) after the addition of TBAF (photo provided by submitters)
After stirring for an additional 3-3.5 h (250 rpm), the reaction is determined to be complete by TLC analysis (Note 32). Aqueous NaOH (1.0 M, 70 mL) (Note 33) is added to the reaction flask, and the mixture is stirred for 10 min at 23 °C. The resulting mixture is transferred to a 1 L separatory funnel (24/40 joint), and is diluted with deionized water (150 mL), saturated aqueous NaCl solution (100 mL), and ethyl acetate (200 mL) (Note 34). The organic and aqueous layers are separated (Note 35). The organic layer is extracted with 0.5 M NaOH (2 x 100 mL), and the combined aqueous layers are washed with ethyl acetate (2 x 100 mL). The aqueous layer is then acidified to pH 2 with 3.0 M HCl (130 mL) (Note 9). The resulting aqueous layer is then extracted with ethyl acetate (3 x 150 mL). The combined organic layers are washed with saturated aqueous NaCl solution (100 mL), dried over Na2SO4 (31 g) (Note 10), filtered through a fritted Büchner funnel (Note 11) into a 1 L round-bottomed collection flask, and concentrated under reduced pressure via a rotary evaporator (Note 36). The resulting residue is dissolved in dichloromethane (10 mL) (Note 37) and purified by flash column chromatography (Note 38). The eluate containing the product (Note 39) is concentrated under reduced pressure (Note 40) on a rotary evaporator which affords benzoic acid (5) as a white solid (3.82 g, 89% yield) (Figure 5) (Notes 41, 42, and 43).

Figure 5. Isolated benzoic acid (5) (photo provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
benzoic acid,
benzoyl chloride,
dichloromethane,
N-methylaniline,
triethylamine,
N-phenyl-N-methyl benzamide,
2-(trimethylsilyl)ethan-1-ol,
bis(cyclooctadiene)-nickel(0),
1,3-bis(2,6-diisopropylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene,
toluene,
tetrabutylammonium fluoride,
tetrahydrofuran,
ethyl acetate,
hexanes,
acetic acid,
sodium hydroxide, and
hydrochloric acid.
2. The desiccator for drying glassware contains silica gel pellets as drying agent. The hot, oven-dried glass apparatus is placed inside the dessicator and cooled to room temperature.
3. The submitters set up the reaction as follows: A single-necked (24/40 joint) 500 mL round-bottomed flask is equipped with a Teflon-coated magnetic stir bar (5.0 x 1.0 cm, cylindrical shaped). The apparatus is flame-dried under reduced pressure and cooled to 23 °C under an atmosphere of nitrogen. The flask is sealed with a rubber septum which is then pierced with a needle connected to a Schlenk line under high vacuum (<1 mmHg), and the flask is evacuated and backfilled with nitrogen (3 cycles). The reaction vessel is then maintained under nitrogen for the duration of the reaction.
4.
Triethylamine (99%) was purchased from Alfa Aesar and passed through an activated alumina column before use. The checkers purchased
triethylamine (> 99.0%) from TCI which was used as received.
5.
N-Methylaniline (
2) (98%) was purchased from TCI and used as received. The checkers purchased
2 (> 98%) from TCI, which was used as received.
6.
Dichloromethane (99.9%) was purchased from Thermo Fisher Scientific and passed through an activated alumina column before use. The submitters transferred
dichloromethane via a cannula. The checkers purchased HPLC grade dichloromethane from RCI Labscan Ltd., which was passed through alumina columns in a PureSolv MD 5 Solvent Purification System before use.
7.
Benzoyl chloride (
1) (99%) was purchased from Millipore Sigma and used as received. The submitters added
1 (4.1 mL, 36 mmol, 1.0 equiv) dropwise via a plastic syringe over 5 min. The checkers purchased
benzoyl chloride (> 99%) from Alfa Aesar, which was used as received. The mass of the
benzoyl chloride used was determined by weighing the
benzoyl chloride-filled syringe before and after injecting 4.1 mL.
8. The progress of the reaction is monitored by TLC analysis on silica gel with 5:1
hexanes:
EtOAc as eluent and visualization with a UV lamp (254 nm). The starting material,
benzoyl chloride (
1) has R
f = 0.67 and the
benzamide product has R
f = 0.20.
Figure 6. TLC of crude reaction mixture after Step A (S = benzoyl chloride, C = co-spot of S and R, and R = reaction mixture) (photo provided by submitters)
9.
Dichloromethane (99.5%) for the workup was purchased from Thermo Fisher Scientific and used as received. The 1.0 M
HCl solution was prepared from concentrated
HCl (36.5-38.0%), which was purchased from Millipore Sigma diluted with deionized water. The checkers purchased AR grade
dichloromethane (99.8%) from RCI Labscan Ltd. which was used as received. The checkers prepared the 1.0 M
HCl solution from diluting with deionized water, concentrated AR grade
HCl (37%) purchased from RCI Labscan Ltd.
10.
NaCl (>99.5%) and
Na2SO4 (>99.0%) were purchased from VWR and Millipore Sigma respectively, and used as received. The checkers purchased
NaCl (AR grade) and Na
2SO
4 (AR grade) from Dieckmann (Hong Kong) Chemical Industry Co. Ltd. which were used as received.
11. Filtration was done using a medium porosity fritted Büchner funnel (150 mL capacity) under vacuum.
12. Rotary evaporation at 30 °C from 350 mmHg to 200 mmHg yielded the crude oil.
13. Careful rotary evaporation at 30 °C, from 450 mmHg to 350 mmHg, followed by drying under high vacuum (<1 mmHg) for 1 h afforded the dry loaded mixture.
14. The OD 8 cm x 45 cm column is wetted using 400 g of silica (SiliaFlash P60, particle size 0.040-0.063 mm, purchased from SiliCycle and used as received) and 750 mL of 5:1
hexanes:
EtOAc. The crude material is dry loaded onto the column, which is then rinsed with three portions of 2 mL of 5:1
hexanes:
EtOAc eluent and the silica column is then covered with a 2 cm thick layer of sand. A gradient of 1650 mL 5:1
hexanes:
EtOAc then 1000 mL 4:1
hexanes:
EtOAc, then 800 mL 3:1
hexanes:
EtOAc, then 600 mL 2:1
hexanes:
EtOAc, and finally 600 mL 1:1
hexanes:
EtOAc was used. The first 1650 mL of eluate is collected into two 1000 mL Erlenmeyer flasks. After this, fraction collection (using 55 mL fractions) begins with the addition of 1000 mL of 4:1
hexanes:
EtOAc. The checkers purchased silica gel (0.040 - 0.063 mm) from Merck KGaA, which was used as received.
15. Fractions containing the product were identified by TLC analysis (using 5:1
hexanes:
EtOAc as eluent). Fractions 18-50 contained the desired product, and each test tube was rinsed with (2 x 1 mL)
EtOAc to ensure quantitative transfer.
16. Rotary evaporation was performed at 30 °C from 150 mmHg to 30 mmHg. The resulting oil was placed under high vacuum (<1 mmHg) for 1 h.
17. The solid is removed from high vacuum and crushed to a powder every 15 min for a total of 2 h under high vacuum.
18. The product is characterized as follows:
1H NMR
pdf (400 MHz, CDCl
3 with TMS) δ: 7.32-7.27 (m, 2H), 7.25-7.19 (m, 3H), 7.18-7.10 (m, 3H), 7.07-7.00 (m, 2H), 3.50 (s, 3H);
13C NMR
pdf (100 MHz, CDCl
3 with TMS) δ: 170.8, 145.1, 136.1, 129.7, 129.3, 128.8, 127.9, 127.0, 126.6, 38.5 ppm; IR (ATR): 3059, 2937, 1634, 1489, 1361, 1299, 1100, 770, 696, 578 cm
-1; HRMS-ESI (
m/z) [M + H]
+calcd for C
14H
14NO
+, 212.1070; found, 212.1070. mp 58 - 60 °C.
19. The purity of
3 was determined to be >97 wt% by qNMR
pdf using 1,3,5-trimethoxybenzene (Sigma-Aldrich, >99.9%) as the internal standard. The purity of
3 was determined to be >99 wt% by checkers via qNMR using 1,3,5-trimethoxybenzene (Sigma-Aldrich, >99.9%) as the internal standard.
20. The checkers performed a duplicate run at full scale which provided 7.1 g (96 % yield) of
3 from 4.1 mL of
benzoyl chloride.
21. After drying in the oven, the condenser was removed from the oven and stoppered with the dried 5 mL protection flask, which was secured by a Keck clip. The other end of the condenser was capped with a septum, and the entire assembly was brought to the fume hood. Immediately, the septum at the top of the reflux condenser was pierced with a needle (18G) connected to an argon manifold and allowed to cool. The protection flask was removed under a positive pressure of dry argon gas. The male joint of the reflux condenser was lightly greased with silicone grease (purchased from Dow Corning), capped with the protection flask again, secured by a Keck clip.
22. An argon-filled glovebox is used to store and load reagents due to the oxygen sensitivity of SIPr and
Ni(cod)2. The submitters used a N
2-filled glovebox.
23. SIPr (98%) was purchased from Strem Chemicals and used as received. The checkers purchased SIPr (98%) from BLD Pharmatech Ltd. and Combi-Blocks, which were used as received.
24.
Ni(cod)2 (98%) was purchased from Strem Chemicals and used as received. The checkers purchased
Ni(cod)2 (98%) from J&K Scientific, which was used as received.
25. The reflux condenser is attached to the reaction flask while in the glovebox to avoid air exposure.
26.
Toluene (99.9%) was purchased from Thermo Fisher Scientific, passed through an activated alumina column, distilled over CaH
2 and freeze-pump-thawed at -196 °C three times to ensure deoxygenation, after which it was stored in the glovebox. The checkers obtained anhydrous and deoxygenated
toluene that was passed through alumina columns under nitrogen in a PureSolv MD 5 Solvent Purification System.
27.
2-(Trimethylsilyl)ethan-1-ol (purity determined by qNMR using 1,3,5-trimethoxybenzene to be 98%) was purchased from Combi-Blocks and was distilled and sparged with nitrogen for 15 min, after which it was stored in the glovebox. The checkers purchased
2-(trimethylsilyl)ethan-1-ol (> 99.0%) from J&K Scientific. The liquid was distilled and then sparged with argon for 1 h.
28. The submitters reported a slightly different procedure, which assembles the reaction entirely inside the glovebox: A jacketed reflux condenser (14/20 joint) equipped with a 5 mL (14/20 joint) round-bottomed flask, and a single-necked (14/20 joint) 250 mL round-bottomed reaction flask equipped with a Teflon-coated magnetic stir bar (4.0 x 1.0 cm, cylindrical-shaped), are flame-dried under reduced pressure and cooled to 23 °C under an atmosphere of nitrogen. The 250 mL reaction flask containing the stir bar is charged with
N-methyl-N-phenyl benzamide (
3) and sealed with a rubber septum. The septum is pierced with a needle connected to a Schlenk line, and the flask is purged with nitrogen for 5 min. The reaction vessel and reflux condenser are then brought into a glovebox. In the glovebox, SIPr and
Ni(cod)2 are added. Then
toluene is added in four portions via a 10 mL pipette. Next,
2-(trimethylsilyl)ethan-1-ol (
4) is added to the mixture. The reaction flask is then fit with the reflux condenser inside the glovebox and brought out of the glovebox. The joint between the flask and condenser is sealed with Teflon tape, and the septum at the top of the reflux condenser is pierced with a needle connected to a Schlenk line under an atmosphere of nitrogen.
29. The progress of the reaction is monitored using TLC analysis on silica gel with 5:1
hexanes:
EtOAc as eluent and visualization with a UV lamp (254 nm). The starting material,
N-methyl-N-phenyl benzamide (
3) has R
f = 0.20 and the ester intermediate has R
f = 0.71.
Figure 7. TLC of crude reaction mixture after Step B, part i (S = amide starting material, C = co-spot of S and R, and R = reaction mixture with ester intermediate) (photo provided by submitters)
30. The submitters connected the addition funnel (24/20) to the reaction flask using an adapter (24/40 to 14/20).
31.
TBAF (0.95-1.10 M in THF with 3.0-7.0% water) was purchased from Sigma Aldrich and used as received. The checkers purchased
TBAF (1.0 M solution in THF) from Energy Chemical, which was used as received.
32. The progress of the reaction is monitored using TLC analysis on a silica gel plate with 5:1
hexanes:
EtOAc as eluent and visualization with a UV lamp (254 nm). The starting material,
N-methyl-N-phenyl benzamide (
3) has R
f = 0.20 and
benzoic acid (
5) has R
f = 0.01.
Figure 8. TLC of crude reaction mixture after Step B part ii (S = amide starting material, C = co-spot of S and R, and R = reaction mixture with benzoic acid) (photo provided by submitters)
33.
NaOH (1.0 M) was prepared by dissolving
NaOH pellets (purchased from Alfa Aesar with 98 % purity) in deionized water. The checkers prepared the 1.0 M
NaOH solution by dissolving
NaOH pellets (AR grade, purchased from Dieckmann (Hong Kong) Chemical Co. Ltd.) in deionized water.
34.
Ethyl acetate (99.5%) was purchased from Thermo Fisher Scientific and used as received. The checkers purchased
ethyl acetate (GR grade) from Duksan Pure Chemicals Ltd. which was used as received.
35. A slight black emulsion forms between the organic layer and transparent aqueous layer. The submitters only collected the clear aqueous layer (see Figure 9).
Figure 9. Reaction mixture after dilution with deionized water, saturated aqueous sodium chloride, and EtOAc (photo provided by submitters)
36. Rotary evaporation at 30 °C from 100 mmHg to 60 mmHg yields the crude solid.
37. The mixture is sonicated to fully dissolve the crude solid.
38. The OD 5.5 cm x 45 cm column is wetted using 250 g of silica and 600 mL of 9:1
hexanes:
EtOAc + 1% AcOH. The crude material is loaded onto the column, and the flask is then rinsed with eluent (3 x 2 mL), which is added to the column. The silica column is then covered with a 2 cm thick layer of sand. Fraction collection (27 mL fractions) begins immediately and an additional 2000 mL of 9:1
hexanes:
EtOAc + 1% AcOH is used.
39. Fractions containing the product were identified by TLC analysis (using 5:1
hexanes:
EtOAc + 1% AcOH as eluent, where
benzoic acid has an R
f = 0.14). Fractions 26-68 contained the desired product, and each fraction is rinsed with (2 x 1 mL)
EtOAc to ensure quantitative transfer.
40. Rotary evaporation is performed at 30 °C from 150 mmHg to 60 mmHg. The product is placed under high vacuum (<1 mmHg) for 1 h resulting in the solidified product.
41. The product is characterized as follows:
1H NMR
pdf (500 MHz, CDCl
3 with TMS) δ: 10.14 (br s, 1H), 8.13 (d,
J = 7.8 Hz, 2H), 7.62 (t,
J = 7.4 Hz, 1H), 7.5 (t,
J = 7.67 Hz, 1H);
13C NMR
pdf (100 MHz, CDCl
3 with TMS) δ: 172.4, 133.8, 130.2, 129.3, 128.5; IR (ATR): 3300-2500 (broad), 2828, 2553, 1679, 1580, 1420, 1288, 928, 700, 666, 546 cm
-1; HRMS-ESI (
m/z) [M + H]
+ calcd for C
7H
7O
2+, 123.0441; found, 123.0442; mp 122.5 - 123 °C.
42. The purity of
5 was determined to be >97 wt% by qNMR
pdf using trimethoxybenzene (Sigma-Aldrich, >99.9%) as the internal standard. The purity of
5 was determined to be >99 wt% by checkers via qNMR using trimethoxybenzene (Sigma-Aldrich, >99.9%) as the internal standard.
43. The checkers performed another run at half-scale which provided 1.69 g (81% yield) of
5.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The procedure described provides a mild means to access carboxylic acids from amide precursors. Carboxylic acids are ubiquitous functional groups found in natural products and drugs but can be difficult to carry through multistep syntheses due to their acidity. Many methods for accessing carboxylic acids proceed from oxidation of alcohols or aldehydes to the corresponding carboxylic acid, but methods to access carboxylic acids from amide precursors are limited. Although hydrolysis can be achieved by subjecting amides to strongly acidic or basic hydrolysis conditions at elevated temperatures, these reaction conditions lack chemoselectivity, particularly in the presence of esters and functional groups containing epimerizable stereocenters. Recent methods to access carboxylic acids from amides include those that rely on the usage of Lewis acid catalysts
2 which coordinate to the amide to promote hydrolysis and also through geometric distortion (i.e., "twisted amides") which interrupt delocalization of the nitrogen lone pair into the amide carbonyl, making hydrolysis more facile as a result of lowered resonance stabilization.
3 Whereas these strategies have proven effective, further developments in this area are coveted in order to achieve mild and chemoselective hydrolysis of a wider range of amide substrates.
Despite the challenges associated with their activation, amides remain attractive functional groups for multistep synthesis due to their inherent stability toward acidic and basic reaction conditions, as well as many redox manipulations. Therefore, in recent years, many groups have sought to develop reaction manifolds to selectively activate and break the amide C-N bond.
4,5,6,7,8 One platform relies on oxidative addition to the amide C-N bond and subsequent cross-coupling of the resulting acyl-metal species (often using
nickel or palladium).
9 Nickel in particular has been shown to be enabling due to its relative ease in participating in oxidative addition.
10 Many cross-coupling transformations of amides have now been explored using carbon, nitrogen, and oxygen-based nucleophiles to access ketones, amides, and esters, respectively.
9 However, using water as a nucleophile for a direct nickel-catalyzed hydrolysis amides to access carboxylic acids was found to be infeasible.
11 The reported one-pot procedure overcomes this challenge by employing silyl alcohol
4 as a nucleophile, resulting in the formation of
6 (Figure 10). Carboxylic acid
5 can then be unveiled in the same pot using
TBAF as a fluoride source.
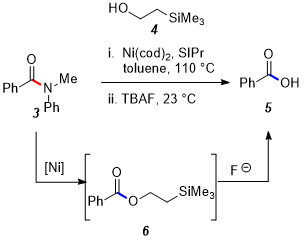
Figure 10. Overview of strategy to access carboxylic acids from amides
As shown with the selected scope in Figure 11, the nickel-catalyzed reaction conditions are tolerant of several different arylamide derivatives. In particular, aryl carboxylic acids with both electron-donating and electron-withdrawing substituents (8c and 8d, respectively) could be formed in good yields from amide precursors. Additionally, the quinoline-containing carboxylic acid 8f could be formed in 60% yield. The methodology also showed promise in its tolerance of sterically and electronically varied amide coupling partners. Despite the bulky i-Pr group on amide 7a, 60% yield of the resulting product, benzoic acid (5), could be observed. The N-tosyl benzamide 7b was also well-tolerated in the reaction and allowed formation of benzoic acid (5) in 71% yield.
Figure 11. Selected scope of carboxylic acids accessible with various aryl-substituents and amide substrates with different N-substituents
This methodology was also shown to be chemoselective and tolerant of epimerizable stereocenters (Figure 12). Valine-derived amide 9 bearing an acidic α-proton, as well as the traditionally more-reactive ester, underwent cross-coupling to yield benzoic acid (5) and secondary amine 10. Importantly, the amine could be isolated with full stereoretention and preservation of the ester moiety. This example further highlights the mildness and chemoselectivity of the method in comparison to other strategies to form carboxylic acids from amides.
<
Figure 12. Chemoselective and stereoretentive amide net-hydrolysis
This procedure provides a simple one-pot approach to access carboxylic acids from amide precursors. Its tolerance toward various aryl and N-substituents and functionalities less stable to traditional hydrolysis methods make it an attractive strategy for accessing carboxylic acids in a comparatively mild and selective manner.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Triethylamine; (121-44-8)
N-Methylaniline; (2) (100-61-8)
Benzoyl chloride; (1) (98-88-4)
SIPr: 1,3-Bis(2,6-di-i-propylphenyl)imidazolidin-2-ylidene; (258278-28-3)
Ni(cod)2: Bis(1,4-cyclooctadiene)nickel(0); (1295-35-8)
2-(Trimethylsilyl)ethan-1-ol; (4) (2916-68-9)
TBAF: Tetrabutylammonium fluoride; (429-41-4)

|
Ana Bulger was born in Concord, MA and raised in Maynard, MA. In 2019, she received her B.S. in Chemistry from Westmont College where she performed research under the supervision of Professor Amanda Silberstein. In 2019, she began her graduate studies at the University of California, Los Angeles in Professor Neil K. Garg's laboratory where she is currently a third-year graduate student. Her research focuses on the activation of amide electrophiles using nickel catalysis as well as on developing synthetic methods using strained intermediates. |

|
Dominick Witkowski was born in Portsmouth, VA and raised in Northborough, MA. In 2020, he received his B.A. in Chemistry from Boston University where he carried out research under the direction of Professor John A. Porco, Jr. In 2020, he began graduate studies at the University of California, Los Angeles, where he is currently a second-year graduate student in Professor Neil K. Garg's laboratory. His studies primarily focus on developing synthetic methods utilizing strained cyclic allenes. |

|
Neil K. Garg is a Distinguished Professor of Chemistry and the Kenneth N. Trueblood Endowed Chair at the University of California, Los Angeles. His laboratory develops novel synthetic strategies and methodologies to enable the total synthesis of complex bioactive molecules. |

|
Cyrus Kwing-Yeung Chan was born in Hong Kong and he obtained his BBiomedSc degree from the University of Hong Kong in 2021. After graduation, he joined the group of Professor Pauline Chiu as a research assistant. He will be pursuing his postgraduate studies in chemistry at Columbia University, New York. |

|
Yu Chen was born in Anhui, Mainland China. She received her B.Sc. in 2013 from Anhui University. In 2020, she completed her Ph.D. at the University of Hong Kong under the supervision of Professor Pauline Chiu. Currently, she is pursuing postdoctoral research at the University of Hong Kong, focusing on organic synthesis and medicinal chemistry. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved