Org. Synth. 2022, 99, 381-400
DOI: 10.15227/orgsyn.099.0381
Transformation of a N,N-Dimethylaniline N-oxide into a Tetrahydroquinoline Scaffold via a Formal Polonovski-Povarov Reaction
Submitted by Elizabeth L. Isaac, Lauren N. Tumbelty, and William J. Chain*
1Checked by Tufan K. Mukhopadhyay and Dirk Trauner
1. Procedure (Note 1)
A. 4-Bromo-N,N-dimethylaniline (2). A 1L single-necked, round-bottomed flask (RBF) equipped with a Teflon-coated magnetic stir bar (12 x 25 mm, oval) is charged with 4-bromoaniline (17.2 g, 99 mmol, 1.0 equiv) (Note 2) followed by sodium cyanoborohydride (31.4 g, 499 mmol, 5 equiv) (Note 3), which is then dissolved in tetrahydrofuran (500 mL) (Note 4) by stirring (800 rpm, under air) for ~5 min at 23-25 °C. Paraformaldehyde (15 g, 499 mmol, 5 equiv) (Note 5) is added to the 1L flask in one portion via funnel (Figure 1A). The flask is then sealed with a rubber septum and placed under nitrogen atmosphere. Glacial acetic acid (30 mL, 499 mmol, 5 equiv) (Note 6) is added dropwise via syringe over 10 min. The resulting mixture is allowed to stir (800 rpm) for 15 min before the reaction flask is put into an IKA heating block that is heated to 50 °C (Figure 1B). The reaction mixture is stirred at that temperature for 24 h. The flask is then removed from the heating block and allowed to cool to room temperature. Excess acetic acid in the resulting mixture is then quenched by slow addition of saturated aqueous sodium bicarbonate (300 mL) (Note 7) and stirred until the vigorous bubbling ceases. The biphasic mixture is diluted with diethyl ether (200 mL) (Note 8) (Figure 1C). Once the layers are separated, the aqueous layer is extracted with diethyl ether (3 x 250 mL) using a 2 L separatory funnel. The organic layers are combined and washed with water (100 mL) and saturated aqueous sodium chloride (100 mL) (Note 9). Anhydrous sodium sulfate (120 g) (Note 10) is then added to the combined organic layers which is allowed to stand for 5 min with occasional swirling. The dried solution is filtered through a 150 mL fritted funnel (24/40) to remove the sodium sulfate (Figure 1D), and the filtered solution is concentrated (40°C, 330 mmHg) on a rotary evaporator to yield a crude white solid (30.2 g) (Note 11) (Figure 1E). This crude material is purified using flash column chromatography on a silica gel column (Note 12) (Figure 1F) using hexanes and ethyl acetate as the eluent (Note 13). Fractions are analyzed by TLC (Note 14), and the fractions containing product are concentrated by rotary evaporation (40 °C, 150 mmHg) to afford 2 (16.29 g, 81%) as a powdery white solid (Notes 15 and 16) (Figure 1G).

Figure 1 (A) Reaction mixture before heating; (B) Reaction mixture under heat; (C) Extraction of aqueous layer from organics; (D) Filtration of sodium sulfate from organics; (E) Crude product after solvent removal; (F) Purification of the crude material via flash chromatography; (F) Pure material as a white powdery solid (Photos provided by checkers)
B. 4-Bromo-N,N-dimethylanline-N-oxide 3-chlorobenzoic acid (3). A 500-mL single-necked, round-bottomed flask (24/40), equipped with a Teflon-coated magnetic stir bar (12x25 mm, oval) is charged with 4-bromo-N,N-dimethylaniline (2) (9.96 g, 49.7 mmol, 1 equiv) followed by m-Chloroperoxybenzoic acid (12.3 g, 54.7 mmol, 1.1 equiv) (Note 17) and is then dissolved in dichloromethane (100 mL) (Note 18) to give a pale yellow solution. The reaction flask is placed in an ice bath (Figure 2A) and the solution is stirred (400 rpm) at 0 °C for 1 h. Basic alumina (39.8 g) (Note 19) is added to the reaction mixture and the resultant slurry is stirred for 15 min. The slurry is filtered through a 150 mL fritted funnel (24/40), and the filtered solution is concentrated (40 °C, 300 mmHg) on a rotary evaporator to yield a beige solid (Figure 2B). The crude solid is re-slurried with ethyl acetate (50 mL) (Note 13) and dried by azeotropic distillation with benzene (3 x 10 mL) (Note 20) to afford 3 (16.41 g, 44.0 mmol, 88%) as a powdery beige solid (Notes 21, 22, and 23) (Figure 2C).

Figure 2. (A) Reaction set-up, (B) Drying of N-oxide salt, (C) Appearance of N-oxide salt (photos provided by checkers)
C. cis-2-Bromo-5-methyl-5,6,6a,11b-tetrahydrobenzofuro[2,3-c]quinoline (4). A 500-mL oven-dried, single-necked round-bottomed flask is charged with 4-bromo-N,N-dimethylaniline-N-oxide 3-chlorobenzoic acid (3) (15 g, 40.3 mmol, 1 equiv) and dissolved in dichloromethane (315 mL) (Note 24) under argon atmosphere. The stirred solution is then charged with di-tert-butyl dicarbonate (Boc2O, 22.8 g, 104.7 mmol, 2.6 equiv) (Note 25), followed by 2,3-benzofuran (12.4 g, 105 mmol, 2.6 equiv) (Note 26) and then cooled to 0 °C by immersion in an ice bath (Figure 3A). After 10 min of stirring (400 rpm, 0 °C) the solution is charged with 4-(Dimethylamino)-pyridine (490 mg, 4.0 mmol, 0.1 equiv) (Note 27). The resulting clear, light orange solution (Figure 3A) is stirred at that temperature for 1 h, whereupon the ice bath is removed and replaced with a dry ice-acetone bath (Figure 3B), further cooling the reaction mixture to -78 °C. After 20 min of stirring (400 rpm, -78 °C), a solution of tin(IV) chloride (1.0 M in dichloromethane, 10.1 mL, 10.06 mmol, 0.25 equiv) (Note 28) is slowly added to the reaction mixture. The resulting amber-brown solution is stirred at -78 °C for 1 h, then the cooling bath is removed, and the solution is allowed to warm slowly to 23 °C and stirred at that temperature for an additional 24 h. The brown reaction mixture (Figure 3C) is then carefully poured over 1M aqueous sodium hydroxide (150 mL) (Note 29) and diluted with 200 mL of dichloromethane (Note 30). The layers are separated, and the aqueous layer is extracted with dichloromethane (1 × 100 mL) in a 2 L separatory funnel (Figure 3D). The combined organic layers are washed sequentially with water (200 mL) and saturated aqueous sodium chloride (200 mL) (Note 31), and the organic layer is dried over anhydrous sodium sulfate (150 g) (Note 32) for 10 min with occasional swirling. The dried solution is then filtered through a 150 mL fritted funnel, and the filtrate is concentrated (40 °C, 300 mmHg) to afford a brown liquid with a pale-yellow precipitate (Figure 3E). The crude material is then purified by flash column chromatography (Note 33) (Figure 3F). Fractions containing the product are identified by TLC analysis (Note 34) are combined and are concentrated (40 °C, 140 mmHg) by rotary evaporator to isolate a pale-yellow powder. The solid is washed with hexane (25 mL) and the clear yellow solution is discarded. Thorough drying under vacuum afforded a pale yellow crystalline solid crystalline solid, which is identified as 4 (10.2 g, 32.3 mmol, 80% yield) as a (Notes 35, 36, and 37) (Figure 1G).

Figure 3. (A) Reaction set up in ice bath (0 ºC); (B) Reaction mixture in dry ice-acetone bath (-78 ºC); (C) Reaction mixture after 24 h of stirring; (D) Extraction of organic layer from aqueous layer; (E) Concentrated crude material; (F) Purification of crude material by column chromatography; (G) Final product after hexane wash (photos provided by checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
4-bromoaniline,
sodium cyanoborohydride,
tetrahydrofuran,
paraformaldehyde,
acetic acid,
sodium bicarbonate,
diethyl ether,
sodium chloride,
sodium sulfate,
hexanes,
ethyl acetate,
meta chloroperoxybenzoic acid,
dichloromethane,
alumina,
benzene,
di-tert-butyl dicarbonate,
2,3-benzofuran,
4-(dimethylamino)pyridine,
tin(IV) chloride, silica gel,
sodium hydroxide,
CDCl3, and
1,3,5-trimethoxybenzene.
2.
4-Bromoaniline was obtained from Oakwood Chemical and used as received.
3.
Sodium cyanoborohydride was obtained from Oakwood Chemical and used as received.
4.
Tetrahydrofuran was purchased from Fisher Chemical and dried by passage through the Pure Solv-MD Standard Design Solvent Purification System from "Innovative Technology. The submitters used the drying procedure described by Pangborn,
et al.
25.
Paraformaldehyde was obtained from Oakwood Chemical and used as received.
6.
Glacial acetic acid was obtained from Fisher Scientific. Addition of
glacial acetic acid must be performed slowly to prevent excessive foaming. In the event of excessive foaming an ice bath can be added.
7.
Sodium bicarbonate was purchased from Fisher Chemical and used as received.
8.
Diethyl ether was purchased from Fisher Chemical and used as received.
9.
Sodium chloride was purchased from Oakwood Chemical and used as received.
10.
Sodium sulfate (anhydrous) was purchased from Oakwood Chemical and used as received.
11. A sticky white solid was observed as the crude product.
12. The crude material is purified by flash chromatography on a silica gel column. The column (5 x 20 cm) was slurry packed with Geduran© Si60 silica gel (40-63 μm) from Merck KGaA (250 g of silica, ~15 cm high) and 100 %
hexanes (
Note 13). The crude product was dissolved in
hexane (30 mL) and
dichloromethane (5 mL) before being added to the column. The column was eluted with 500 mL
hexanes:
ethyl acetate (
Note 13) (98:2) followed by 500 mL
hexanes:
ethyl acetate (95:5), 1 L
hexanes:
ethyl acetate (90:10), and 1.5 L
hexanes:
ethyl acetate (85:15). The desired product was obtained in fractions 32-66 (50 mL fractions were collected) (
Note 14).
13.
Hexanes was purchased from Fisher Chemical and used as received. Ethyl acetate was purchased from Fisher Chemical and used as received.
14. The fractions should be checked by TLC analysis (Silica gel F
254 glass backed TLC plates from Merck KGaA were used) with
hexanes:
EtOAc (3:2) as eluent. Product R
f=0.75, impurities present at ~0.85, ~0.30 and ~0.10.
Figure 4. TLC plate under 254 nm UV light (SM-starting material; Co-spot; C-Crude). Impurities may not be visible when viewed under UV light, but cerium ammonium molybdate staining is more effective in revealing the presence of impurities (photo provided by submitters)
15.
4-Bromo-N,N-dimethylaniline (
2): mp 53-55 °C;
1H NMR (400 MHz,
CDCl3) δ: 7.31 (d,
J = 9.1 Hz, 2H), 6.59 (d,
J = 9.1 Hz, 2H), 2.93 (s, 6H).
13C NMR (101 MHz,
CDCl3) δ: 149.6, 131.8, 114.2, 108.6, 40.7. FTIR (ATR) 3091, 2925, 1866, 1591, 1354 cm
-1. HRMS (ESI) Calcd. for C
8H
11BrN [(M
79Br+H)
+]: 200.0069 and [(M
81Br+H)
+]: 202.0049. Found: 200.0081 and 202.0059. Purity was assessed by quantitative
1H NMR
pdf using 20 mg (100 µmol) of 2 and 16.8 mg (100 µmol) of
1,3,5-trimethoxybenzene dissolved in 0.5 mL
CDCl3. Integrating the
N,N-dimethyl resonance against the trimethoxy signal showed 99% purity.
16. A second reaction performed on identical scale provided 16.1 g (81%) of the same product.
17.
m-Chloroperoxybenzoic acid (
mCPBA) was obtained from Sigma Aldrich (77%). It was used as received utilizing the indicated purity level.
18.
Dichloromethane (unstabilized HPLC grade) was purchased from Fisher Chemical and purified by the method of Pangborn,
et al.
2 19. Basic
alumina was purchased from Acros Organics and used as received.
20.
Benzene was purchased from Alfa Aesar and purified by the method of Pangborn,
et al.
221. The product may also be yellow or turn yellow over time.
4-Bromo-N,N-dimethylanline-N-oxide 3-chlorobenzoic acid (
3): mp 115-116 °C,
1H NMR (400 MHz,
CDCl3) δ: 8.04 (t,
J = 1.9 Hz, 1H), 7.94 (dt,
J = 7.7, 1.4 Hz, 1H), 7.84 (d,
J = 9.1 Hz, 1H), 7.64 (d,
J = 9.1 Hz, 1H), 7.45 (ddd,
J = 7.9, 2.2, 1.1 Hz, 1H), 7.33 (t,
J = 7.9 Hz, 1H), 3.83 (s, 6H)
13C NMR (101 MHz, CDCl
3) δ: 169.3, 151.8, 135.2, 134.2, 132.9, 131., 130.0, 129.5, 127.9, 124.0, 121.7, 62.1. FTIR (ATR): 3572, 3032, 1659, 1647, 1567, 1070 cm
-1. HRMS (ESI): Calcd. for C
8H
11BrNO [(M
79Br+H)
+]: 216.0019. Found: 216.0019.
22. Purity was assessed by quantitative
1H NMR
pdf using 20 mg (53.7 µmol) of
3 and 9.04 mg (53.7 µmol) of
1,3,5-trimethoxybenzene were dissolved in 0.5 mL
CDCl3. Integrating the
N,N-dimethyl resonance against the trimethoxy signal showed 90% purity.
23. A second reaction on the same scale provided 15.4 g (83%) of the same product
24.
Dichloromethane (unstabilized HPLC grade) was purchased from Fisher Chemical and dried by passage through the Pure Solv-MD Standard Design Solvent Purification System from Innovative Technology. The submitters used the drying procedure described by Pangborn,
et al.
225.
Di-tert-butyl dicarbonate (
Boc2O) was purchased from Oakwood Chemical and used as received.
26.
2,3-Benzofuran was purchased from Oakwood Chemical and used as received.
27.
4-(Dimethylamino)-pyridine was purchased from Oakwood Chemical and used as received.
28.
Tin (IV) chloride solution (1.0 M in
dichloromethane) was purchased from Sigma Aldrich and used as received.
29.
Sodium hydroxide was purchased from Fisher Chemical. To prepare the 1M solution used in this reaction,
sodium hydroxide (10 g, 250 mmol) was dissolved in 250 mL of distilled water.
30.
Dichloromethane was purchased from Fisher Chemical and used as received.
31.
Sodium chloride was purchased from Oakwood Chemical and used as received.
32.
Sodium sulfate (anhydrous) was purchased from Oakwood Chemical and used as received.
33. The crude material is purified by flash chromatography on a silica gel column. The column (5 x 20 cm) was slurry packed with Geduran© Si60 silica gel (40-63 μm) from Merck KGaA (250 g of silica, ~15 cm high) using
hexanes (100 mL) (
Note 13). The crude product was dissolved in minimal
dichloromethane (15-20 mL) before being added to the column. The column was eluted with 1.5 L
hexanes:
ethyl acetate (
Note 13) (98:2), followed by 2.5 L
hexanes: ethyl acetate (95:5). The desired product is obtained in fractions 43-72 (50 mL fractions were collected) (
Note 34).
34. The fractions were checked by TLC analysis (Silica gel F
254 glass-backed TLC plates from Merck KGaA were used)) with
hexanes:
EtOAc (9:1) as eluent. Product R
f=0.40, impurities possible at ~0.90, ~0.75 , ~0.50, ~0.30 and ~0.05.
Figure 5. TLC plate under 254 nm UV light (left) and cerium ammonium molybdate staining (right). (SM BF: benzofuran (undistilled), CR: crude reaction mixture, PDT: known product reference spot, SM 3: 4-bromo-N,N-dimethylaniline-N-oxide, 3-chlorobenzoic acid salt). The impurities may not be visible in UV, but cerium ammonium molybdate staining shows the impurities listed above (Photos provided by checkers)
35.
cis-2-Bromo-5-methyl-5,6,6a,11b-tetrahydrobenzofuro[2,3-c]quinoline (
4): mp 123.6-124.8 °C,
1H NMR (400 MHz,
CDCl3) δ: 7.45 (d,
J = 2.3 Hz, 1H), 7.23 (dd,
J = 8.7, 2.4 Hz, 1H), 7.19 (d,
J = 7.5 Hz, 1H), 7.11 (t,
J = 7.7 Hz, 1H), 6.83 (td,
J = 7.5, 1.0 Hz, 1H), 6.79 (d,
J = 8.0 Hz, 1H), 6.55 (d,
J = 8.7 Hz, 1H), 5.26 (dt,
J = 9.5, 3.6 Hz, 1H), 4.61 (d,
J = 9.4 Hz, 1H), 3.33 (dd,
J = 12.5, 3.8 Hz, 1H), 3.12 (dd,
J = 12.5, 3.4 Hz, 1H), 2.81 (s, 3H).
13C NMR (101 MHz,
CDCl3) δ: 159.4, 147.1, 131.0, 130.4, 129.8, 128.8, 127.3, 125.0, 120.9, 114.3, 110.96, 109.7, 81.8, 54.6, 42.9, 39.7. FTIR (cm
-1): 3046, 3040, 2957, 1596, 1318, 1241, 1236 (
Note 38). HRMS (ESI): Calcd. for C
16H
15BrNO [(M
79Br+H)
+]: 316.0332 and [(M
81Br+H)
+]: 318.0311. Found: 316.0287 and 318.0303.
36. Purity was assessed by quantitative
1H NMR
pdf using 20 mg (63.3 µmol) of 4 and 10.6 mg ( 63.3 µmol) of
1,3,5-trimethoxybenzene in 0.5 mL
CDCl3. Integrating the
N,N-dimethyl resonance against the trimethoxy signal showed 99.8% purity.
37. A second reaction on the same scale provided 10.1 g ( 80%) of the same product.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Tetrahydroquinolines are among the most privileged scaffolds in organic chemistry, composing the basis of many alkaloids, biologically active natural products, medicinal materials, catalysts, ligands, and other critically useful structures.
3,4 There are a number of strategies toward the synthesis of tetrahydroquinolines including the hydrogenation of quinolines,
5 tandem hydroamination-hydrogenation of alkynyl anilines,
6 and the classical Povarov reaction
7 which combines an aniline, an aldehyde, and some alkene component in a formal inverse-electron demand aza-Diels-Alder
8 reaction (Scheme 1). Each of these approaches is quite powerful, but all of them can be limited by the nature of the reaction conditions and the reagents required. The hydrogenation of quinolines is achieved by exposure to hydrogen gas and various transition metals
9 or organocatalysts,
10 and despite extensive investigations and progress in these areas, these reduction conditions are often still extreme and the preparation of quinoline starting materials is not trivial. The tandem hydroamination-hydrogenation of alkynyl anilines is conducted via metal catalysis
6 and can be combined with hydrogenation; however this route is limited in scope to 2-substituted products by virtue of the alkyne starting material (substituent R
2). The classical Povarov reaction is initiated by the condensation of an aniline and an aldehyde, typically with some kind of acid catalyst; the resultant aromatic imine constitutes an
aza-diene that can undergo a stepwise cationic capture with an alkene. As a result, the classical Povarov reaction is at the mercy of electron-rich condensation partners, and suitable alkene reaction participants that can survive the conditions required to generate the aromatic
aza-diene. Each of these strategies supports asymmetric synthesis, however these methods further limit the scope of the available substrates by requiring structural features that facilitate catalysis.
11
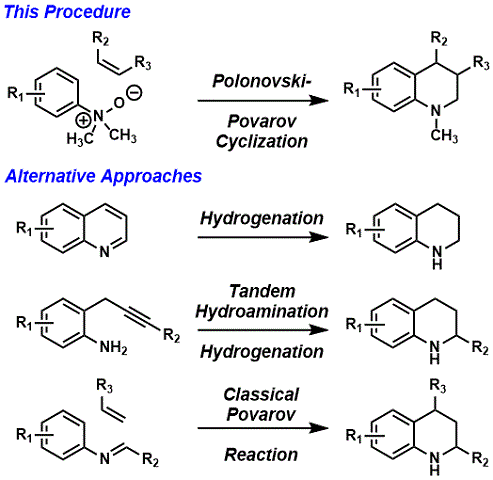
Scheme 1. Approaches to Tetrahydroquinolines
We were attracted to the tetrahydroquinoline nucleus, the opportunities afforded by the Povarov reaction,
7 and the formal inverse-electron demand
aza-Diels-Alder reaction
8 that takes place on the aromatic platform. While the condensation event in a traditional Povarov reaction can limit the scope to electron-rich participants, we felt we could intercept reactive iminium ions by exploiting reactivity we had previous sought to avoid in our work with aniline-
N-oxides. We have been interested in the wide array of functionalizations that are facilitated by the oxidation of anilines to the corresponding aniline-
N-oxides.
12,13,14,15 Classical aromatic substitution chemistries are problematic with electron-rich anilines,
16 however by rendering the nitrogen lone pair an
N-
O bond we have unlocked a host of functionalization reactions that give access to a wide array of substituted anilines. We have described protocols for the oxygenation,
12 alkylation,
12 and halogenation
13 of
N,
N-dialkylanilines, as well as successive oxidation cycles of
N,
N-dialkylanilines to give 3-carboalkoxyindolines.
14 While the mechanisms of these functionalization reactions can be ambiguous, one liability associated with all of these chemistries is a Polonovski-type elimination
17 involving the
N-
O bond to give an iminium ion electrophile. However, this liability presents the opportunity to form new bonds by nucleophilic capture of the iminium ion by a suitable nucleophile. We have described protocols by which this capture can occur in both intra- and intermolecular settings, but when the nucleophilic participant can support a cation, the formal
aza-Diels-Alder reaction is realized (Scheme 2).
15
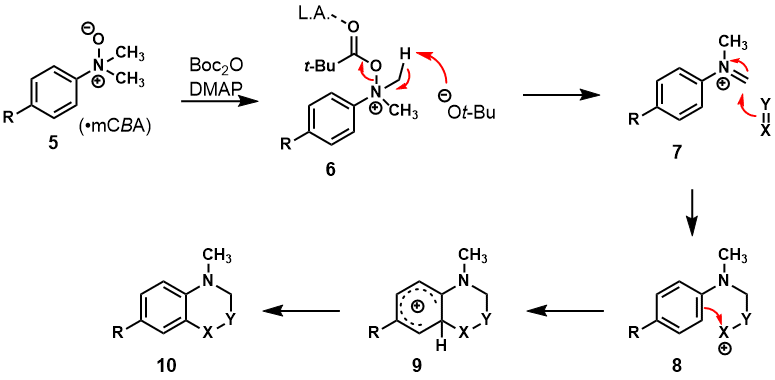
Scheme 2. Mechanism of the tandem Polonovski−Povarov reactions with aniline N-oxides
The iminium ion (7) is functionally equivalent to the intermediates generated in the classical Povarov reaction, but is generated without the problematic condensation event. Thus, cyclizations were achieved with a variety of nucleophilic alkenes to give a diverse array of tetrahydroquinolines, and these cyclization reactions are robust and amenable to scale. Thus far in our studies, we have generated a host of such products by treatment of N,N-dialkylaniline-N-oxides with six different groups of nucleophilic reaction partners (Scheme 3).
Scheme 3. Scope of formal Polonovski-Povarov reactions with N,N-dimethylaniline-N-oxides
Appendix
Chemical Abstracts Nomenclature (Registry Number)
4-Bromoaniline: benzenamine; (586-77-6)
Sodium cyanoborohydride: Sodium borocyanohydride; (25895-60-7)
Paraformaldehyde: Polyoxymethylene; (30525-89-4)
Glacial acetic acid: Acetic acid; (64-19-7)
meta-Chloroperoxybenzoic acid: Benzenecarboperoxoic acid; (937-14-4)
4-(Dimethylamino)pyridine: 4-Pyridinamine (1122-58-3)
2,3-Benzofuran: (271-89-6)
Tin (IV) Chloride : Tin (IV) Chloride Solution; (7646-78-8)

|
Elizabeth Isaac obtained a B.S. in Chemistry in 2018 from Otterbein University where also conducted research in the laboratory of Prof. Robin Grote investigating cyclodehydration chemistry. She is currently pursuing her Ph.D. degree in the Chain Group in the Department of Chemistry & Biochemistry at the University of Delaware. |

|
Lauren Tumbelty obtained a B.S. in Biological Sciences in 2016 from Rowan University. She then obtained an M.S. in Pharmaceutical Sciences from Rowan University conducting research in the laboratory of Prof. Gustavo Moura-Letts investigating heterocycle synthesis via dipolar cycloaddition chemistries. Lauren is currently is pursuing a Ph.D. degree in the Chain Group in the Department of Chemistry & Biochemistry at the University of Delaware. She is also a fellow of the Chemistry Biology Interface (CBI) Program at UD. |

|
William Chain obtained a B.S. in Chemistry in 2001 from Pennsylvania State University. In 2006, he received his Ph.D. under the guidance of Professor Andrew G. Myers at Harvard University. After post-doctoral studies with Professor Erik J. Sorensen at Princeton University, he was appointed Assistant Professor in 2009 at the University of Hawai'i. In 2015, he was appointed Associate Professor in the Department of Chemistry & Biochemistry at the University of Delaware. His research interests include the development of new reactions utilizing aromatic platforms, synthetic methods for the construction of small and medium sized rings, and natural products synthesis. |

|
Tufan K. Mukhopadhyay received his B.Sc. degree in India. Then he obtained his M.Sc. degree from Indian Institute of Technology Madras in 2010. Thereafter, he studied base metal catalysis during his Ph. D. at Arizona State University under the supervision of Prof. Ryan J. Trovitch. Following his Ph.D. in 2016, and after conducting two years of postdoctoral research, he joined the research group of Prof. Dirk Trauner at New York University as a postdoctoral associate to study synthesis of lipids, bioactive natural products and explore photopharmacology of small azobenzene molecules. His research interests include sustainable catalysis, synthesis of bioactive natural products, and medicinal chemistry. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved