Org. Synth. 2025, 102, 381-397
DOI: 10.15227/orgsyn.102.0381
Synthesis of meta-Substituted Benzenes via Mo-Catalyzed Intermolecular Deoxygenative Coupling of Ynones and Allylic Amines
Submitted by Yi-Zhe Yu, Gang Fang, Hong-Yi Su, and Chun-Xiang Zhuo*
1Checked by Yanyao Liu and Kevin Campos
1. Procedure (Note 1)
A. 1,8-bis-(Diphenylphosphino)octane (DPPO). An oven-dried, 100-mL Schlenk flask containing a magnetic stir bar (1.0 x 0.5 cm, Teflon-coated) (Note 2) and a rubber septum was flushed with nitrogen (Note 3). The flask was cooled to -78 ℃ and stirred at this temperature (22-25 ℃). To the flask were added dry THF (20 mL) (Note 4) and subsequently n-BuLi (2 M in hexane, 12 mL, 24 mmol, 2.4 equiv) (Note 5) via syringe. After 5 min, diphenylphosphine (3.72 g, 3.5 mL, 20 mmol, 2.0 equiv) (Note 6) was added dropwise by a syringe. The reaction mixture was stirred at -78 ℃ for 20 min. Then 1,8-dibromooctane (2.72 g, 1.8 mL, 10 mmol, 1.0 equiv) (Note 7) was added dropwise into the lithium diphenylphosphide solution using a syringe while the solution was stirred at -78 ℃. The reaction mixture was gradually warmed to room temperature (22-25 ℃) and stirred at this temperature for 10 h under N2 atmosphere. Then the reaction mixture was quenched with 5 mL of saturated aq. NaHCO3 solution, diluted with 50 mL of brine and extracted with CH2Cl2 (150 mL and 50 mL × 2). The combined organic layers were dried over anhydrous Na2SO4 (20 g for 15 min), filtered and concentrated by a rotary evaporator (200 mbar down to 3 mbar) to provide the crude mixture as a white solid (Note 8). The mixture was purified by flash column chromatography (Note 9) on silica gel to obtain 1,8-bis(diphenylphosphino)octane (4.4 g, 91%) (Note 10) as a white powder in more than 99% purity, as determined by quantitative 1H NMR spectroscopy (Note 11).

Figure 1. A. Reaction setup for the synthesis of DPPO. B. The reaction mixture after quenching (Photo provided by authors)
B. 3-Phenyl-1-(p-tolyl)prop-2-yn-1-one (1). To an oven-dried, 250-mL Schlenk flask containing a magnetic stir bar (1.0 x 0.5 cm, Teflon-coated) (Note 2) and a rubber septum, Pd(PPh3)2Cl2 (175 mg, 0.25 mmol, 0.5 mol %) (Note 12) and CuI (95 mg, 0.5 mmol, 1 mol%) (Note 13) were added under N2 atmosphere (Note 3). To the flask were subsequently added dry THF (50 mL) and triethylamine (50 mL) (Note 4) by syringe. After cooling the mixture in an ice-water bath for 5 min, the p-toluoyl chloride (7.73 g, 6.6 mL, 50 mmol, 1.0 equiv) (Note 14) and phenylacetylene (5.21g, 5.6 mL, 51 mmol, 1.02 equiv) (Note 15) were added dropwise to the solution via syringe. The reaction mixture was continually stirred for 5 h. During this period, the ice-bath was allowed to naturally return to room temperature (22-25 ℃). Then, the reaction mixture was transferred into a round-bottomed flask (250 mL). The reaction flask was washed with CH2Cl2 (5 mL × 3). The combined solvents were concentrated by a rotary evaporator (200 mbar down to 3 mbar) to afford about 20 mL of the mixture, and then 50 mL of EtOAc was added (Note 16). The mixture was filtered, and the residue was washed with 10 mL of EtOAc three times. The organic layers were combined, and then the solvents were removed by a rotary evaporator (200 mbar down to 3 mbar) to provide the crude product as a brown solid. The crude product was purified by column chromatography on silica gel (Note 17) and then recrystallized from petroleum ether (Note 18) to provide product 1 (10.2 g, 93%) (Note 19) as a light-yellow solid in 99% purity, as determined by quantitative 1H NMR spectroscopy (Note 20).

Figure 2. A. Reaction setup for the synthesis of compound 1; B. The crude reaction mixture before column chromatography (Photo provided by authors)
C. 4-Methyl-1,1':3',1''-terphenyl (3). A 50 mL EasyMax SS316 reactor (vessel 1, Note 21) (Figure 3) with an overhead pitched-blade impeller, pressure gauge, gas inlet and outlet, and thermocouple in thermowell was charged under nitrogen with molybdenum hexacarbonyl (317 mg, 10 mol %, 1.20 mmol) (Note 22) and 3,5-di-tert-butylcyclohexa-3,5-diene-1,2-dione (264 mg, 10 mol %, 1.20 mmol) (Note 23). Then, 20 mL of dry toluene (Note 4) was added by a syringe and the solution was dark red-purple and homogeneous. The vessel was evacuated for 5 sec to -10 psig then backfilled to ~0 psig with nitrogen. This backfill/purge procedure was repeated five times, and then the vessel was sealed, heated to 160 ℃, and held for 15 min with stirring (500 rpm). The vessel was then cooled to room temperature with stirring (500 rpm) and light nitrogen flow. In a 400-mL EasyMax vessel (Vessel 2) outfit as above was charged under nitrogen with compound 1 (6.34 g, 2.40 equiv, 28.8 mmol) and DPPO (2.89 g, 0.50 equiv, 6.00 mmol). The solution from vessel 1 was transferred via syringe to EasyMax Vessel 2). Vessel 1 was rinsed with additional toluene (20 mL) and transferred into Vessel 2 via syringe. Additional toluene (80 mL) was added to Vessel 2 followed by N-allylmethylamine (889 mg, 1.20 mL, 96% wt, 1.00 equiv, 12.0 mmol) via syringe (Note 24 and Note 25). Vessel 2 was sealed and was heated to 160 ℃ and held for 48 h with stirring.

Figure 3. EasyMax Reactor used in Step C (Photo provided by checkers)
After 48 h, the reaction mixture was cooled to room temperature (22-25 ℃) and transferred into a round-bottomed flask (250 mL). The reaction tube was washed with CH2Cl2 (5 mL × 3). The combined solvents were evaporated by a rotary evaporator (200 mbar down to 3 mbar) to give the crude mixture, which was purified by flash column chromatography on silica gel (Note 26) to afford the title compound 3 as a white solid (2.16 g, 74%) (Note 27) in 99% purity, as determined by quantitative 1H NMR spectroscopy (Note 28).
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of “Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also “Identifying and Evaluating Hazards in Research Laboratories” (American Chemical Society, 2015) which is available via the associated website “Hazard Assessment in Research Laboratories” at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
Mo(CO)6, as well as the proper procedures for heating in a closed system under the temperature above the boiling point of the solvent.
2. All glassware and Teflon-coated magnetic stir bar were thoroughly washed and dried in an oven at 110 ℃ for at least 5 h.
3. This operation was performed by evacuating the tube and backfilling with
N2 three times via a Schlenk line.
4. The solvents were purified by distillation over the following drying agents and were transferred under argon or
N2 atmosphere:
THF (Na),
triethylamine (
CaH2),
toluene (Na). The checker used brand new sure-sealed solvents from Sigma-Aldrich.
5.
n-BuLi was purchased from Sigma-Aldrich (sealed, 2 M in
hexane, light yellow solution) and used as received under positive pressure of
nitrogen atmosphere. The checker used 2.5 M
n-BuLi in
hexane from Sigma-Aldrich. The checker also found 2.0 M
n-BuLi in cyclohexane did not work in their hands.
6.
Diphenylphosphine (colorless liquid) was purchased from Thermo Scientific and used as received.
7.
1,8-Dibromooctane (97% purity, colorless oil) was purchased from TCI and used as received.
8. The extraction and concentration operations should be performed as fast as possible because the
DPPO solution was easily oxidized.
9. The product was purified by flash column chromatography on a column (46 × 254 mm) with 85 g of silica gel (300-400 mesh). The crude product in 20 mL of
CH2Cl2/
petroleum ether (v/v = 1/1) was loaded and eluted with 400 mL of
CH2Cl2/
petroleum ether (v/v = 1/1).
10. The desired product was obtained from fractions 6 to 10, each tube contained about 30 mL of eluent. The combined eluents were concentrated by a rotary evaporator at 40 ℃ (water bath) and dried under vacuum (1.0 mm Hg) for 2 h to give 4.40 g (91%) of
DPPO as a white powder with >99 % purity as determined by qNMR
pdf with 1,3,5-trimethoxybenzene as and internal standard.
11. Characterization data for
DPPO:
1H NMR
pdf (400 MHz, CDCl
3): δ = 7.52 - 7.40 (m, 8H), 7.39 - 7.27 (m, 12H), 2.14 - 2.01 (m, 4H), 1.52 - 1.36 (m, 8H), 1.33 - 1.18 (m, 4H).
13C NMR
pdf (100 MHz, CDCl
3): δ = 139.0 (d,
J P-C = 13.0 Hz), 132.7 (d,
J P-C = 18.3 Hz), 128.4 (s), 128.3 (d,
J P-C = 6.6 Hz), 31.1 (d,
J P-C = 12.9 Hz), 29.0 (s), 28.0 (d,
J P-C = 11.2 Hz), 25.9 (d,
J P-C = 15.9 Hz).
31P NMR
pdf (162 MHz, CDCl
3): δ = -16.1.
12.
Pd(PPh3)2Cl2 (yellow powder) was purchased from Sigma-Aldrich and used as received.
13.
CuI (98% purity, gray powder) was purchased from Sigma-Aldrich and used as received.
14.
p-Toluoyl chloride (98% purity, light yellow liquid) was purchased from Sigma-Aldrich and used as received.
15.
Phenylacetylene (98% purity, light yellow liquid) was purchased from Sigma-Aldrich and used as received.
16. A large amount of white powder precipitated after this operation.
17. The product was purified by flash column chromatography on a column (46 × 254 mm) with 85 g of silica gel (300-400 mesh). The crude product was loaded with 20 mL of
petroleum ether/
CH2Cl2 (v/v = 1:1) and eluted with 1 L of
petroleum ether/
EtOAc (v/v = 20:1). The authors were able to get 99 wt % pure after a single chromatography and following recrystallization. When the checker repeated this reaction, the last a few fractions coeluted with an impurity which required a second chromatography.
18. The desired product was obtained from fractions 11 to 27, each tube contained about 30 mL eluent. The combined eluents were concentrated by a rotary evaporator at 40 ℃ (water bath).
19. The obtained product with a small amount of impurities was recrystallized from 20 mL of
n-hexane and dried under vacuum (1.0 mmHg) for 2 h to give 10.2 g (93%) of compound
1 as a light-yellow powder with >99 % purity as determined by qNMR
pdf with 1,3,5-trimethoxybenzene as and internal standard. Procedure for recrystallization: To a 100 mL round-bottomed flask are added the crude product and 20 mL
n-hexane. The suspension is heated to reflux for 30 min until all the solids are dissolved. Cool the flask to room temperature and place it in a refrigerator at -20 ℃ for 2 h. The solids are then filtered using a filter paper on a Buchner funnel after they are thoroughly precipitated. About 5-10 mL cooled
n-hexane could be used to wash the resulting solids which are then collected and dried under vacuum for 2 h.
20. Characterization data for compound
1:
1H NMR
pdf (400 MHz, CDCl
3): δ = 8.16 - 8.09 (m, 2H), 7.72 - 7.65 (m, 2H), 7.50 - 7.45 (m, 1H), 7.45 - 7.38 (m, 2H), 7.34 - 7.28 (m, 2H), 2.44 (s, 3H).
13C NMR
pdf (100 MHz, CDCl
3): δ = 177.7, 145.2, 134.5, 133.0, 130.6, 129.6, 129.3, 128.6, 120.2, 92.6, 86.9, 21.8.
21. The authors utilized a two-vessel procedure with 100-ml and 250-ml Young Schlenk flasks equipped with magnetic stir bars. (Caution: a protective shield should be installed around the Schlenk tube due to the risk of explosion during the reaction).
Figure 4. A. Toluene solution of Mo(CO)6 and 3,5-di-tert-butyl-1,2-benzoquinone before heating; B. Toluene solution of Mo(CO)6 and 3,5-di-tert-butyl-1,2-benzoquinone after heating; C. Tube B with substrate and DPPO; D. The reaction mixture after 48 h (Photo provided by authors)
22.
Mo(CO)6 (98 % purity, white powder) was purchased from Alfa Aesar and used as received.
23.
3,5-Di-tert-butyl-1,2-benzoquinone (99% purity, purple black powder) was purchased from TCI and used as received.
24. According to the mechanism of this reaction (See Discussion section),
methylamine was produced during the reaction. Therefore, excess of ynone
1 was proposed for the capture of
methylamine.
25.
N-Allylmethylamine (96% purity, colorless liquid) was purchased from Sigma-Aldrich and distilled under
N2 atmosphere from
KOH before use. The authors assumed it's still 96% purity after distillation and the volume charged here is after purity correction. The Checker utilized a brand-new ampule that was opened right before use and no distillation over
KOH was performed.
26. The product was purified by flash column chromatography on a column (46 × 254 mm) with 85 g of silica gel (300-400 mesh). The crude product was mixed with 15 g of silica gel (300-400 mesh) in
CH2Cl2 (100 mL), and the resulting mixture was concentrated by a rotary evaporator to dryness and loaded to the column which was eluted with 1.1 L of
petroleum ether.
27. The desired product was obtained from fractions 14 to 35, each tube contained about 30 mL eluent. The combined eluents were concentrated by a rotary evaporator at 40 ℃ (water bath) and dried under vacuum (1.0 mm Hg) for 2 h to give 2.16 g (74%) of
3 as a white solid with >99% purity as determined by qNMR
pdf with 1,3,5-trimethoxybenzene as and internal standard.
28. Characterization data for compound for compound
3:
1H NMR
pdf (400 MHz, CDCl
3): δ = 7.86 (t,
J = 1.5 Hz, 1H), 7.73 - 7.68 (m, 2H), 7.65 - 7.58 (m, 4H), 7.57 - 7.49 (m, 3H), 7.45 - 7.40 (m, 1H), 7.33 (d,
J = 8.0 Hz, 2H), 2.46 (s, 3H).
13C NMR
pdf (100 MHz, CDCl
3): δ = 141.7, 141.6, 141.2, 138.2, 137.2, 129.5, 129.1, 128.8, 127.3, 127.2, 127.1, 125.92, 125.9, 125.8, 21.1.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Substituted benzene derivatives occupy a prominent position among ring systems utilized extensively in organic synthesis, material science, and medicinal chemistry. Their significance in drug discovery is underscored by their ubiquitous presence within the top 200 best-selling small molecule pharmaceuticals.
2 (Figure 4). Nevertheless, a recent study into the distribution of benzenoid substitution patterns in approved small molecule drugs has revealed that 1,3-di- and 1,3,5-tri-substituted benzene derivatives are significantly less common compared to other substitution patterns (including 1-; 1,2-; 1,4-; and 1,2,4-).
3 This disparity may stem from the scarcity of robust, efficient, and user-friendly synthetic methodologies specifically designed to target these derivatives.
Figure 5. Selected examples of bioactive molecules containing meta-substitued benzenes.
Traditional methods for synthesizing 1,3-di- and 1,3,5-tri-substituted benzene derivatives often involve electrophilic aromatic substitution of benzene derivatives with deactivating substituents
4, as well as transition-metal-catalyzed [2 + 2 + 2] cyclotrimerization of alkynes
5 (Scheme 1A). However, these approaches often encounter limitations such as low reactivity and limited product diversity. More recent endeavors have centered on utilizing directing groups
6 or norbornene-mediated
7 transition-metal-catalyzed
meta-C-H functionalization strategies, steric or electronic effects induced Ir- or Pd-catalyzed
meta-C-H borylation
8a,b or olefination
8c reactions, Pd-catalyzed dehydrogenative [3 + 3] aromatization
9, and oxidative NHC-catalyzed benzene formation.
10 Nevertheless, these methods frequently necessitated the use of directing groups, precious metals, specific substrates, or excessive amounts of oxidants and bases. Consequently, there is a pressing need to devise an efficient, versatile, convenient, and non-noble metal-catalyzed approach for constructing 1,3-di- and 1,3,5-tri-substituted benzene derivatives, particularly those bearing unsymmetric and unfunctionalized alkyl or aryl groups that are challenging to access.
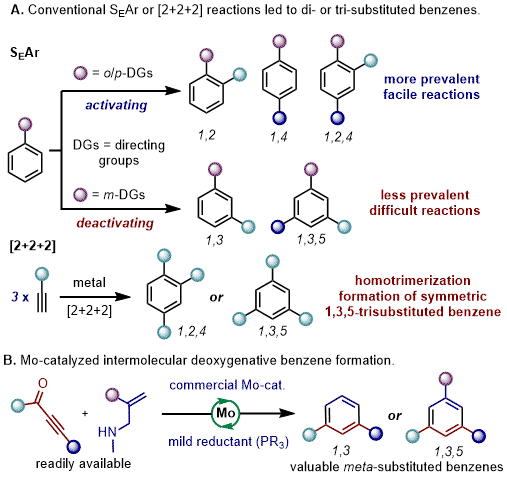
Scheme 1. Methods for the syntheses of substituted benzenes
Recently, we reported a general and efficient molybdenum-catalyzed intermolecular deoxygenative benzene forming reaction (Scheme 1B).
11 This novel strategy provided a straightforward access to a series of 1,3-di and 1,3,5-trisubstituted benzene derivatives from readily available starting materials (ynones and allylamines). The substrate scope of the reaction was broad. For example, as shown in Figure 5, several
meta-substituted benzene products bearing unsymmetric and unfunctionalized alkyl or aryl groups were easily prepared from the corresponding ynones and allylic amines. Moreover, this method was successfully applied in the derivatization of a series of biologically active molecules.
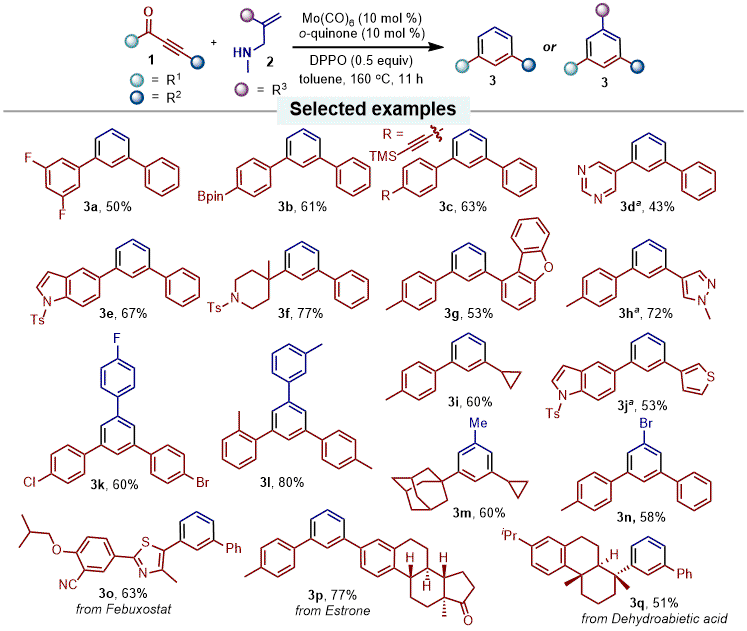
Figure 6. Selected examples of the substrate scope for Mo-catalyzed intermolecular deoxygenative benzene-forming reaction. Reaction conditions: Mo(CO)6 (10 mol %), o-quinone (10 mol %), 1 (0.48 mmol, 2.4 equiv), 2 (0.2 mmol, 1.0 equiv), and DPPO (0.1 mmol, 0.5 equiv) in toluene at 160 ℃. aThe reaction was performed with 20 mol % of Mo(CO)6/o-quinone for 5 h on a 0.1 or 0.2 mmol scale
Preliminary mechanistic studies (Figure 6) suggested that the low-valent molybdenum catalyst species M-1 in-situ formed by Mo(CO)6 and quinone catalyzed the intermolecular 1,4-aza-Michael addition reaction between allylamine 2 and ynone 1 to form Int-1 which then underwent [1,5]-hydride shift, cyclization, and retro-hydroamination to produce meta-substituted benzene product 3. During this process, the catalytically active species M-1 was regenerated from the reduction reaction of oxo-Mo-species M-3 byphosphine.
Figure 7. A plausible reaction pathway
In summary, the novel molybdenum-catalyzed intermolecular deoxygenative reaction that transformed readily accessible ynones and allylic amines into benzene derivatives offered a straightforward and modular route to synthesize a diverse array of unsymmetric and unfunctionalized 1,3-di- and 1,3,5-tri-substituted benzene compounds, many of which were previously challenging to obtain. Notably, the molybdenum catalyst effectively orchestrates multiple reaction processes, including aza-Michael addition, [1,5]-hydride shift, cyclization, and aromatization, demonstrating its remarkable efficiency and versatility. We anticipate that this method will enrich the arsenal of medicinal chemists in their pursuit of drug discovery.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
1,8-Dibromooctane; (4549-32-0)
Diphenylphosphine; (829-85-6)
n-Butyllithium; (109-72-8)
p-Toluoyl chloride; (874-60-2)
Phenylacetylene; (536-74-3)
Bis(triphenylphosphine)palladium(II) dichloride; (13965-03-2)
Cuprous iodide; (7681-65-4)
N-Allylmethylamine; (627-37-2)
Molybdenum hexacarbonyl; (13939-06-5)
3,5-di-tert-Butyl-1,2-benzoquinone; (3383-21-9)

|
Chun-Xiang Zhuo received his BSc in chemistry from Hunan University in 2009. He completed his PhD at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (SIOC) in 2014 under the supervision of Prof. Shu-Li You before doing postdoctoral studies with Prof. Alois Fürstner at Max-Planck-Institut für Kohlenforschung. In April 2019, he joined the Department of Chemistry at Xiamen University as a Professor to start his independent research. His current research interests include development of novel synthetic methodology, asymmetric catalysis, natural product synthesis, and medicinal chemistry. |

|
Yi-Zhe Yu was born in Anhui, China, in 1996. He received his BSc degree in 2017 from Anhui Normal University. He is now performing PhD studies under the supervision of Prof. Chun-Xiang Zhuo's laboratory at the College of Chemistry and Chemical Engineering at Xiamen University. His research interests include development of novel molybdenum catalysis and molybdenum catalysts. |

|
Gang Fang was born in Zhejiang, China. He graduated from Xiamen University in 2022 with a BSc degree in Chemical Biology. He is continuing his graduate studies at the Xiamen University under the supervision of Professor Chun-Xiang Zhuo. He is currently focused on the development of novel molybdenum catalysis. |

|
Hong-Yi Su was born in Hunan, China. He is doing his undergraduate studies at the Department of chemistry at Xiamen University. He is currently focused on the development of novel molybdenum catalysis. |

|
Yanyao Liu was born in Chongqing, China. He received his bachelor degree in Chemistry from Nankai University. He then moved to the US and obtained his M.S. at Rutgers University. In 2024, he graduated from Indiana University where he finished his Ph.D. research on boron enable photosensitized [2+2]-cycloadditions. Upon graduation, he joined Merck Research Laboratories and worked in small molecule process chemistry. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved