Org. Synth. 1978, 58, 24
DOI: 10.15227/orgsyn.058.0024
BORANES IN FUNCTIONALIZATION OF DIENES TO CYCLIC KETONES: BICYCLO[3.3.1]NONAN-9-ONE
Submitted by Bruce A. Carlson
1 and Herbert C. Brown
2.
Checked by J. C. Bottaro and G. Büchi.
1. Procedure
Caution! The oxidation with 30% hydrogen peroxide in the last step of Part B may become vigorous and exothermic. No difficulties have been encountered under the conditions described; however, the oxidation should be carried out in a hood behind a protective shield.
A. Lithium triethylcarboxide (Solution 1). A thoroughly dried, 1-l., three-necked, round-bottomed flask fitted with a septum inlet with serum cap, a reflux condenser, and a magnetic stirrer is purged with and maintained under an atmosphere of dry nitrogen. After a solution of 300 ml. of 1.67 M n-butyllithium (0.500 mole) in hexane (Note 1) is introduced into the flask by syringe (Note 2) and cooled to 0° in an ice bath, 58 g. (0.50 mole) of 3-ethyl-3-pentanol (Note 3) is added slowly, but constantly, by syringe (Note 4). At the end of addition the yellow tint of the n-butyllithium solution disappears. The alkoxide solution is standardized by hydrolysis of aliquots in water and by titration of the resulting lithium hydroxide with standard acid to a phenolphthalein end point (Note 5).
B. Bicyclo[3.3.1]nonan-9-one (2). A thoroughly dried, 3-l., three-necked, round-bottomed flask fitted with a serum cap and a magnetic stirrer under nitrogen is charged with 42.3 g. (0.347 mole) of 9-borabicyclo[3.3.1]nonane (9-BBN) (Note 6) before 500 ml. of anhydrous tetrahydrofuran (Note 7) is added using a double-ended syringe needle (Note 2). The flask is fitted with a dry, 500-ml., pressure-equalizing dropping funnel while the flask is purged with a rapid stream of dry nitrogen. The apparatus is maintained under a static pressure of nitrogen throughout the reaction. A solution of 42.3 g. (0.347 mole) of 2,6-dimethylphenol (Note 8) in 75 ml. of anhydrous tetrahydrofuran is added slowly by syringe, and the mixture is stirred at room temperature for 3 hours. Once hydrogen evolution is complete (Note 9), 350 ml. of a 1.46 M solution of 1 in hexane is introduced into the dropping funnel after purging it with dry nitrogen. The clear solution in the flask is cooled to 0°, and 44 g. (35 ml., 0.38 mole) of dichloromethyl methyl ether (Note 10) is added. Solution 1 is then added slowly over approximately 30 minutes. The ice bath is removed, and the flask is warmed to room temperature for 90 minutes (Note 11). A heavy white precipitate of lithium chloride forms (Note 12), and the nitrogen atmosphere is no longer required. A solution of 300 ml. of 95% ethanol, 70 ml. of water, and 42 g. of sodium hydroxide is added, and the mixture is cooled to 0° with efficient stirring. Oxidation is carried out over a 40–45 minute period by the slow, dropwise addition of 70 ml. of 30% hydrogen peroxide (Note 13), while the temperature is maintained below 50° with a cooling bath. After addition the mixture is heated with stirring to 45–50° for 2 hours (Note 14) and then cooled to room temperature. Water (300 ml.) is added, and the aqueous phase is saturated with sodium chloride. The organic phase is separated and washed with 100 ml. of saturated aqueous sodium chloride. The solvents are removed on a rotary evaporator, and the resulting orange liquid is diluted with 500 ml. of pentane, which is then extracted with 250-ml. and 100-ml. portions of aqueous 3 M sodium hydroxide to remove 2,6-dimethylphenol. After washing with 100 ml. of saturated aqueous sodium chloride, the pentane is removed on a rotary evaporator. The 3-ethyl-3-pentanol is removed by distillation under a water aspirator vacuum, b.p. 54–56° (16 mm.) (Note 15). The resulting semisolid residue is dissolved in 200 ml. of pentane and filtered to remove the impurities. Ketone 2 is crystallized by cooling the filtrate to −78°. Ketone 2 is collected by suction filtration, washed with 50 ml. of −78° pentane (Note 16), and dried (Note 17), yielding 35–40 g. (78–83%) of pure ketone 2, m.p. 154–156° (Note 18). Evaporation of the filtrate to ca. 50 ml. and cooling to −78° gives another 3–4 g. of ketone 2, m.p. 149–154°. The total yield of bicyclo[3.3.1]nonan-9-one (2) is 38–44 g. (79–91%).
2. Notes
1.
n-Butyllithium in hexane was obtained from Foote Mineral Company. The solution can be titrated for total alkyllithium by the procedure of Watson and Eastham,
3 and for total base by hydrolysis of aliquots and titration against standard acid prior to use.
n-Butyllithium solution of good purity is essential for success of the reaction.
2.
A double-ended syringe needle is most convenient for transfer of reagents and handling of air-sensitive solutions. Transfer is completed by applying
nitrogen pressure. See references
4 and
5,6,7,8,9 for descriptions of this procedure and for general techniques for handling air-sensitive reagents.
3.
3-Ethyl-3-pentanol (triethylcarbinol) (98%) was purchased from Chemical Samples Company, 469 Kenny Road, Columbus, Ohio 43221, and used without further purification. It is also available from Aldrich Chemical Company, Inc.4.
Addition causes vigorous reaction with evolution of
butane and heat. Redissolution of the
butane in the solution may create a partial vacuum, causing air to be sucked back into the flask; the addition should be kept at such a rate to prevent this. The solution may reflux, but this causes no harm.
5.
Alternatively, if only one use of the solution of
lithium triethylcarboxide (1) is planned, the required amounts of
butyllithium solution and
3-ethyl-3-pentanol may be reacted, and the resulting solution of
lithium triethylcarboxide used without standardization.
6.
9-Borabicyclo[3.3.1]nonane (9-BBN) was purchased from Aldrich Chemical Company, Inc., and used directly. Alternatively, 9-BBN may be prepared by hydroboration of
1,5-cycloöctadiene.
5,6,7,8,9 Caution! 9-BBN is air sensitive and can be pyrophoric.7.
Tetrahydrofuran, containing less than 0.002% peroxides, supplied by the William A. Mosher, Ph.D. Company, 101 Townsend Road, Newark, Delaware 19711, was used directly.
Tetrahydrofuran from other sources may require purification prior to use. Stirring with
lithium aluminum hydride or
sodium benzophenone ketyl, followed by distillation under an inert atmosphere, is the recommended procedure for removal of water and peroxides (see
reference 4, p. 256).
8.
2,6-Dimethylphenol (99%), from Chemical Samples Company, was used without purification. It is also available from Aldrich Chemical Company, Inc.9.
Addition causes evolution of
hydrogen and is carried out over
ca. 10 minutes to prevent foaming. The undissolved 9-BBN rapidly goes into solution. Completion of the reaction may be measured by monitoring
hydrogen evolution with a gas meter. The theoretical amount of
hydrogen is 7.8 l. at STP.
10.
Dichloromethyl methyl ether (98%) was purchased from Aldrich Chemical Company, Inc. (listed as
α,α-dichloromethyl methyl ether) and used without purification. Better results can be obtained if the material is distilled under
nitrogen prior to use. Unlike
chloromethyl methyl ether and
bis(chloromethyl) ether, dichloromethyl methyl ether is reported to have no significant carcinogenic activity;
10 however, as a precaution it should be handled carefully in a well-ventilated hood.
11.
Longer reaction times (
e.g., allowing the reaction to stir overnight) will not affect the product.
12.
The checkers observed no precipitate in their two runs. On occasion the submitters have also noted no
lithium chloride precipitate during a run or sudden precipitation of the salt. Apparently the
lithium chloride supersaturates on these occasions.
13.
The
30% hydrogen peroxide was obtained from Fisher Scientific Company.14.
Initial heating should be done cautiously in case an additional exotherm is experienced.
15.
3-Ethyl-3-pentanol may be removed rapidly by condensation into an
acetone–dry ice cold trap.
16.
The temperature of the solution should be kept as close as possible to −78° during filtration to prevent losses of the first crop of product. On humid days crystallization and filtration are best accomplished in a closed system to prevent excessive condensation of water.
17.
The product has a high vapor pressure and sublimes readily. Care should be taken in drying so that losses of product are not incurred. Drying briefly in a
vacuum desiccator over Drierite is recommended.
18.
Literature,
11 m.p.
155–158.5°; the
2,4-dinitrophenylhydrazone was prepared by the standard procedure and recrystallized from
ethyl acetate–
ethanol to provide orange prisms, m.p.
190–191.2° lit.,
11 m.p.
191.8–192.3°.
3. Discussion
Dialkylborinic acids are conveniently prepared by the hydroboration of olefins with monochloroborane diethyl etherate
7 or, in some cases, by controlled hydroboration of two equivalents of olefin
7 with
borane–
tetrahydrofuran or
borane–
methyl sulfide complex followed by alcoholysis. The base-induced reaction of
dichloromethyl methyl ether with borinic acid esters provides a route to a variety of ketones
12 and olefins.
13,14 The reaction presumably proceeds
via generation of a haloalkoxy carbanion or carbene which reacts rapidly with the borinic acid ester, resulting in transfer of the alkyl groups from boron to carbon. The α-chloroboronic ester intermediate, thus generated, is exceptionally versatile. Oxidation results in the formation of the corresponding ketone in high yield,
12,15 whereas pyrolysis or solvolysis with aqueous
silver nitrate14 provides the internal olefin:
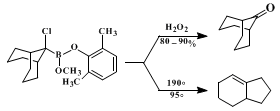
Lithium triethylcarboxide (1) is the base of choice for these conversions. The use of less hindered alkoxide or amide bases results in poorer yields. In this procedure 1.5–2.0 equivalents of base are required, although with more bulky alkyl groups attached to boron only 1 equivalent is necessary. The use of the more hindered 2,6-diisopropylphenol, forming the borinic ester, gives a 96% yield of the bicyclic ketone with only 1 equivalent of base; however, in the work-up procedure this phenol is more difficult to separate from the ketone.
Carbonylation of trialkylboranes in the presence of water
9 and the cyanation of thexyldialkylboranes
16 offer alternative routes to ketones from organoboranes. Neither procedure can utilize dialkylborinic esters in the synthesis and, thus, result in loss of one alkyl group. Carbonylation requires a moderate pressure of
carbon monoxide in some cases, and the cyanation reaction involves the use of strongly electrophilic reagents; neither route has been used successfully in the preparation of
bicyclo[3.3.1]nonan-9-one (2). Previous synthetic routes to this interesting bicyclic ketone
17 have generally required numerous steps and resulted in low overall yields.
11,18 The best alternative procedure, which gives a 60% yield of the bicyclic ketone, involves reaction of
nickel carbonyl with
1,5-cyclooctadiene.
19The base-induced reaction of
dichloromethyl methyl ether with trialkyl- and triarylboranes also provides a powerful method for the preparation of the corresponding tertiary carbinols.
20,21 In this case, all three groups transfer readily from boron to carbon under mild conditions, and oxidation with alkaline
hydrogen peroxide provides the tertiary alcohol.
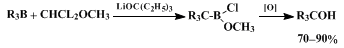
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2,4-dinitrophenylhydrazone
sodium benzophenone ketyl
Lithium triethylcarboxide
bis(chloromethyl) ether, dichloromethyl methyl ether
ethanol (64-17-5)
ethyl acetate (141-78-6)
hydrogen (1333-74-0)
sodium hydroxide (1310-73-2)
carbon monoxide (630-08-0)
sodium chloride (7647-14-5)
silver nitrate (7761-88-8)
nitrogen (7727-37-9)
hydrogen peroxide (7722-84-1)
Pentane (109-66-0)
phenolphthalein (77-09-8)
chloromethyl methyl ether (107-30-2)
borane (7440-42-8)
methyl sulfide (75-18-3)
butane (106-97-8)
butyllithium,
n-butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
nickel carbonyl
3-ethyl-3-pentanol,
Triethylcarbinol (597-49-9)
lithium aluminum hydride (16853-85-3)
hexane (110-54-3)
Lithium chloride (7447-41-8)
Dichloromethyl methyl ether,
α,α-dichloromethyl methyl ether (4885-02-3)
2,6-dimethylphenol (576-26-1)
1,5-cyclooctadiene
lithium hydroxide (1310-65-2)
Bicyclo[3.3.1]nonan-9-one (17931-55-4)
9-borabicyclo[3.3.1]nonane (280-64-8)
2,6-diisopropylphenol (2078-54-8)
bicyclo[3.3.1]nonane (280-65-9)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved