Org. Synth. 1976, 55, 122
DOI: 10.15227/orgsyn.055.0122
SECONDARY AND TERTIARY ALKYL KETONES FROM CARBOXYLIC ACID CHLORIDES AND LITHIUM PHENYLTHIO(ALKYL)CUPRATE REAGENTS: tert-BUTYL PHENYL KETONE
[1-Propanone, 2,2-dimethyl-1-phenyl]
Submitted by Gary H. Posner
1 and Charles E. Whitten.
Checked by Joyce M. Wilkins and Herbert O. House.
1. Procedure
Caution! tert-Butylithium is extremely pyrophoric and must not be allowed to come into contact with the atmosphere. This reagent should only be handled by individuals trained in its proper and safe use. It is recommended that transfers be carried out by using a 20-mL or smaller glass syringe filled to no more than 2/3 capacity, or by cannula. For a discussion of procedures for handling air-sensitive reagents, see Aldrich Technical Bulletin AL-134. [Note added August 2009]
Caution! Since the odor of the thiophenol (benzenethiol) used in this preparation is unpleasant, both steps of this preparation should be conducted in a hood and the glassware used should be washed before it is removed from the hood.
A. Lithium phenylthio(tert-butyl)cuprate. A dry, 200-ml., round-bottomed flask is fitted with a magnetic stirring bar and a 100-ml., pressure-equalizing dropping funnel, the top of which is connected to a nitrogen inlet. After the apparatus has been flushed with nitrogen, 50 ml. of 1.60 M (0.080 mole) n-butyllithium (Note 1) solution is placed in the flask and cooled with an ice bath. Under a nitrogen atmosphere, a solution of 8.81 g. (0.0801 mole) of freshly distilled thiophenol (Note 2) in 30 ml. of anhydrous tetrahydrofuran (Note 3) is added dropwise to the cooled, stirred solution. An aliquot of the resulting solution (Note 4) is standardized by quenching in water, followed by titration with 0.10 N hydrochloric acid to a green end point with a bromocresol indicator. The concentration of lithium thiophenoxide prepared in this manner is typically 1.0 M.
A dry, 250-ml., three-necked round-bottomed flask is equipped with a sealed mechanical stirrer (Note 5), a glass stopper, and a rubber septum through which are inserted hypodermic needles used to evacuate the flask and to admit nitrogen. After the apparatus has been flushed with nitrogen, 4.19 g. (0.0220 mole) of purified copper(I) iodide (Note 6) is added, and while warming with a flame, the apparatus is evacuated, then refilled with nitrogen. After this procedure has been performed twice, the flask is allowed to cool, the stopper is replaced with a thermometer, and 45 ml. of anhydrous tetrahydrofuran is added (Note 3) with a hypodermic syringe. With continuous stirring, 22 ml. of 1.0 M (0.022 mole) lithium thiophenoxide solution is added with a syringe to the slurry of copper(I) iodide. After 5 minutes, the resulting yellow solution is cooled, with continuous stirring, to −65° with an acetone–dry ice cooling bath. Some copper(I) thiophenoxide usually separates from solution at ca. −45°. When the temperature of the mixture has reached ca. −65°, 13.6 ml. (0.0218 mole) of 1.60 M tert-butyllithium (Note 7) solution is added with a syringe to the stirred mixture at such a rate that the temperature of the mixture remains at −60° to −65°. The resulting cloudy yellow-orange solution of the cuprate reagent is stirred at −60° to −65° for 5 minutes (Note 8).
B. tert-Butyl phenyl ketone. With a syringe a solution of 2.81 g. (0.0200 mole) of freshly distilled benzoyl chloride (Note 9) in 15 ml. of anhydrous tetrahydrofuran (Note 3) is added dropwise, with stirring, to the cold solution (−60° to −65°) of the cuprate reagent. The resulting yellow-brown solution is stirred for 20 minutes (at −60° to −65°) and quenched by the addition, with a syringe, of 5 ml. of anhydrous methanol. The red-orange reaction mixture is allowed to warm to room temperature and then poured into 100 ml. of aqueous saturated ammonium chloride. The copious precipitate of copper(I) thiophenoxide is separated by suction filtration and washed thoroughly with several 50-ml. portions of diethyl ether. The combined filtrate is extracted with three 100-ml. portions of ether. The combined ethereal solution is washed with two 50-ml. portions of aqueous 1 N sodium hydroxide and with one 50-ml. portion of aqueous 2% sodium thiosulfate. Each of the aqueous washes is extracted in turn with a fresh 50-ml. portion of ether. The combined ethereal solution is dried with anhydrous magnesium sulfate, filtered, and concentrated by distillation through a short Vigreux column. The residual pale yellow liquid (Note 10) is distilled through a short column under reduced pressure, yielding 2.73–2.82 g. (84–87%) of tert-butyl phenyl ketone as a colorless liquid b.p. 105–106° (15 mm.), 114–115° (44 mm.), n20D 1.5092, n25D 1.5066 (Note 11).
2. Notes
1.
Solutions containing approximately
1.6 M n-butyllithium in hexane were purchased either from Alfa Inorganics, Inc., or from Foote Mineral Company. The concentration of
n-butyllithium in these solutions can be determined either by a double titration procedure
2 or by dilution with anhydrous
tetrahydrofuran, followed by titration with
2-butanol3 in the presence of a
2,2'-bipyridyl indicator [
Org. Synth., Coll. Vol. 6 121 (1988)]. In either case the total base concentration in the reagent is determined by titration with standard aqueous acid.
2.
Thiophenol, purchased from Aldrich Chemical Company, Inc., was redistilled before use; b.p.
65–66° (42 mm.).
3.
Commercial anhydrous
tetrahydrofuran was distilled from
lithium aluminum hydride and stored under
nitrogen.
4.
The submitters report that this solution may be stored under
nitrogen at 0° for several days without deterioration.
Phenylthiocopper is now commercially available from the Alpha Division of the Ventron Corp., 152 Andover St., Danvers, Massachusetts 01923.
5.
Although the submitters had recommended use of a magnetic stirring bar, the checkers encountered considerable difficulty in maintaining adequate stirring of the cold reaction mixture with a magnetic stirrer and recommend use of a sealed mechanical stirrer such as a
Truebore® stirrer.
6.
Copper(I) iodide, purchased from Fisher Scientific Company, was purified by continuous extraction with anhydrous
tetrahydrofuran in a
Soxhlet extractor for approximately 12 hours, to remove colored impurities. The residual
copper(I) iodide was then dried under reduced pressure at 25° and stored under
nitrogen in a
desiccator.
7.
A
pentane solution of tert-butyllithium (purchased from either Alfa Inorganics, Inc., or Lithium Corporation of America, Inc.) was standardized by one of the previously described titration procedures
(Note 1). If possible, it is desirable to use a freshly opened bottle of
tert-butyllithium since previously used bottles of this reagent often contain
lithium tert-butoxide, which will lead to formation of a contaminant in the final product
(Note 10).
8.
Although the submitters report that this reagent is stable at 0° (
i.e., still reactive toward
benzoyl chloride) for periods of at least one hour under a
nitrogen atmosphere,
3,4 the checkers repeatedly observed evidence of thermal decomposition when the solution was allowed to warm above −40°. This decomposition was indicated by the appearance of a red-brown coloration as the reagent was warmed to −40°; as the temperature was raised further to −25° and to 0°, the mixture progressively exhibited a darker brown color.
9.
Benzoyl chloride (purchased from Eastman Organic Chemicals) was redistilled before use; b.p.
35–36° (0.5 mm.).
10.
The checkers found that with previously opened bottles of
tert-butyllithium, the crude product was often contaminated with
tert-butyl benzoate (from lithium tert-butoxide; see (Note 7)). The presence of this impurity in the crude product may be detected either by the presence of an extra IR peak at 1720 cm.
−1 (conjugated ester), or by GC analysis. On a 1.3-m. GC column, packed with silicone fluid, No. SE-52, suspended on Chromosorb P and operated at 155°, the retention time of
tert-butyl phenyl ketone was 4.4 minutes, and the retention times of potential impurities,
methyl benzoate and
tert-butyl benzoate were 2.4 minutes and 7.8 minutes, respectively. If a small amount of
tert-butyl benzoate is present in the crude product, it is most easily removed by heating a mixture of the crude product with
1% by weight of p-toluenesulfonic acid on a
steam bath for 10 minutes followed by partitioning the product between
ether and aqueous
sodium hydrogen carbonate. After the resulting ether solution has been dried and distilled, pure
tert-butyl phenyl ketone is obtained.
11.
The product exhibits a single GC peak (see
(Note 10)). The spectral properties of the product are as follows; IR (CCl
4) cm.
−1: 1680 (conjugated ketone), 1395 and 1370 [C(CH
3)
3];
1H NMR (CCl
4), δ(multiplicity, number of protons, assignment): 1.30 [s, 9H, C(C
H3)
3], 7.2–7.9 (m, 5H, C
6H
5); UV (95% C
2H
5OH) nm. max. (ε): 237 (7350) and 272 (620); mass spectrum
m/e (relative intensity): 162 (M,45), 106 (28), 105 (100), 77 (63), 57 (40), 51 (23), and 41 (30). The physical constants reported for the product are: b.p.
103–104° (13 mm.).
5 n20D 1.5090.
6
3. Discussion
tert-Butyl phenyl ketone has been prepared by the reactions of
benzoic acid with
tert-butyllithium,
7 of
acetophenone with
methyl iodide and base,
5,9 of
benzaldehyde with
tert-butylmagnesium chloride followed by oxidation,
10, and of
2,2-dimethylpropanoyl chloride with
phenylmagnesium bromide.
11
The procedure described here illustrates the preparation of mixed lithium arylhetero(alkyl)cuprate reagents and their reactions with carboxylic acid chlorides.
4 These mixed cuprate reagents also react with α,α'-dibromoketones,
12 primary alkyl halides,
4 and α,β-unsaturated ketones,
4 with selective transfer of the alkyl group.
Two limitations on the utility of organocopper reagents have often been the difficulty in using thermally unstable lithium
sec- and especially
tert-alkylcuprates
13 and the need for a large (
e.g., 300–500%) excess of an organocuprate to achieve complete conversion of substrate to product. Both of these limitations are circumvented by using lithium phenylthio(
tert-alkyl)cuprates, which react with approximately equimolar amounts of carboxylic acid chlorides, forming the corresponding
tert-alkyl ketones in high yield, even with the yield based on the transferred alkyl group. Furthermore, this alkyl group transfer can be achieved in the presence of other functional groups (
e.g., remote halogen or ester functionalities) in the carboxylic acid chloride substrate (Equation 1). Transfer of secondary alkyl groups can also be accomplished efficiently in this way (Equation 2).
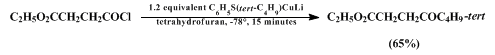
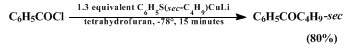
The reaction of
tert-alkyl Grignard reagents with carboxylic acid chlorides in the presence of a
copper catalyst provides
tert-alkyl ketones in substantially lower yields than those reported here.
4,14 The simplicity and mildness of experimental conditions and isolation procedure, the diversity of substrate structural type, and the functional group selectivity of these mixed organocuprate reagents render them very useful for conversion of carboxylic acid chlorides to the corresponding secondary and tertiary alkyl ketones.
15This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
hydrochloric acid (7647-01-0)
methanol (67-56-1)
ether,
diethyl ether (60-29-7)
ammonium chloride (12125-02-9)
sodium hydroxide (1310-73-2)
sodium hydrogen carbonate (144-55-8)
sodium thiosulfate (7772-98-7)
nitrogen (7727-37-9)
Benzoic acid (65-85-0)
copper (7440-50-8)
benzaldehyde (100-52-7)
Acetophenone (98-86-2)
benzoyl chloride (98-88-4)
Methyl iodide (74-88-4)
Phenylmagnesium bromide (100-58-3)
methyl benzoate (93-58-3)
Pentane (109-66-0)
Thiophenol,
Benzenethiol (108-98-5)
copper(I) iodide (7681-65-4)
magnesium sulfate (7487-88-9)
bromocresol
butyllithium,
n-butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
lithium aluminum hydride (16853-85-3)
hexane (110-54-3)
2-Butanol (78-92-2)
2,2'-bipyridyl (366-18-7)
p-toluenesulfonic acid (104-15-4)
1-Propanone, 2,2-dimethyl-1-phenyl,
tert-Butyl phenyl ketone (938-16-9)
lithium thiophenoxide
copper(I) thiophenoxide (34012-88-9)
Phenylthiocopper
2,2-dimethylpropanoyl chloride (3282-30-2)
tert-Butyllithium (594-19-4)
tert-butylmagnesium chloride (677-22-5)
lithium tert-butoxide (1907-33-1)
tert-butyl benzoate (774-65-2)
Lithium phenylthio(tert-butyl)cuprate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved