Org. Synth. 1995, 72, 21
DOI: 10.15227/orgsyn.072.0021
DIASTEREOSELECTIVE HOMOLOGATION OF D-(R)-GLYCERALDEHYDE ACETONIDE USING 2-(TRIMETHYLSILYL)THIAZOLE: 2-O-BENZYL-3,4-ISOPROPYLIDENE-D-ERYTHROSE
[1,3-Dioxolane-4-acetaldehyde, 2,2-dimethyl- α-(phenylmethoxy)-, [R-(R*,R*)]-]
Submitted by Alessandro Dondoni and Pedro Merino
1.
Checked by Gregory P. Brengel and Albert I. Meyers.
1. Procedure
A. 2-(Trimethylsilyl)thiazole (2-TST). A 500-mL, four-necked, round-bottomed flask, containing a magnetic stirring bar, is equipped with two 100-mL, pressure-equalizing dropping funnels and a low-temperature thermometer (Note 1). The dry apparatus is filled with argon and kept under a slightly positive pressure (ca. 100 mm) of this gas for the entire reaction time. The flask is charged with 200 mL of freshly distilled diethyl ether (Note 2) and 111 mL of a 1.5 M solution of butyllithium (0.167 mol) in hexane (Note 3) and (Note 4). One of the two dropping funnels is charged with 25 g (0.152 mol) of 2-bromothiazole (Note 5) in 50 mL of diethyl ether (Note 2) and the other with 16.5 g (0.152 mol) of chlorotrimethylsilane (Note 6) in 50 mL of diethyl ether (Note 2). The reaction flask is cooled to −78°C in a dry ice-acetone bath. While the solution in the flask is stirred, 2-bromothiazole is added dropwise over a period of 1 hr. After 20 min additional stirring, chlorotrimethylsilane is added dropwise over 30 min and stirring is continued for 1 hr at −78°C. The resulting mixture is then allowed to warm up to room temperature. A saturated aqueous sodium bicarbonate solution (200 mL) is added and the mixture is transferred into a 1-L separatory funnel. After the mixture is shaken, the organic layer is recovered and the aqueous layer is extracted with diethyl ether (200 mL). The combined organic layers are dried over anhydrous sodium sulfate, filtered, and concentrated at reduced pressure by rotary evaporation with the external bath temperature not higher than 40°C. The residue is distilled from a 100-mL flask at reduced pressure in a Claisen apparatus equipped with a 10-cm Vigreux column (Note 7). The distillation, after a forerun at 40–65°C (15 mm) consisting mainly of bromobutane, gives 20.2 g (85%) of 2-(trimethylsilyl)thiazole as a colorless liquid, bp 88–91°C (16–17 mm) (Note 8).
B. (1R)-2,3-Di-O-isopropylidene-1-(2-thiazolyl)-D-glycitol. A 100-mL, three-necked, round-bottomed flask, containing a magnetic stirring bar, is equipped with a 50-mL pressure-equalizing dropping funnel and a low-temperature thermometer (Note 1). The dry apparatus is filled with argon and kept under an inert gas pressure of ca. 100 mm for the entire reaction time. The flask is charged with 2.0 g (0.0154 mol) of D-glyceraldehyde acetonide (Note 9) in 25 mL of dichloromethane (Note 10) and the dropping funnel is filled with 2.4 g (0.0153 mol) of 2-TST in 25 mL of dichloromethane (Note 10). The reaction flask is cooled to 0°C in an ice bath. While the solution is stirred, 2-TST is added dropwise over a period of 15 min. After the reaction is stirred for 12 hr at room temperature, it is complete as shown by TLC (silica, diethyl ether – petroleum ether 1:1). Dichloromethane (ca. 40 mL) is removed under reduced pressure and the residue is treated with tetrabutylammonium fluoride (14 mL of a 1.1 M solution in tetrahydrofuran, 0.0154 mol) (Aldrich Chemical Company, Inc.) Desilylation is complete as shown by TLC (silica, diethyl ether – petroleum ether, 1:1) in 10 min. Volatile material is removed under reduced pressure and the residue is treated with 50–60 mL of water. The mixture is extracted with dichloromethane (3 × 50 mL) and the combined dichloromethane solutions are dried with anhydrous sodium sulfate. The solvent is removed under reduced pressure and the solid residue (3.14 g) is flash chromatographed (silica, diethyl ether – petroleum ether, 3:2) to give 2.85 g (84%) of the alcohol as a white solid, mp 114–116°C (from dichloromethane-hexane) (Note 11).
C. (1R)-O-Benzyl-2,3-di-O-isopropylidene-1-(2-thiazolyl)-D-glycitol. A 250-mL, two-necked, round-bottomed flask containing a magnetic stirring bar, is equipped with a 50-mL pressure-equalizing dropping funnel. The dry apparatus is filled with argon and kept under an inert gas pressure of ca. 100 mm greater than the atmosphere until the aqueous work-up (Note 1). The flask is charged with 2.6 g (0.012 mol) of the alcohol (from Step B) in 100 mL of tetrahydrofuran (Note 12) and the dropping funnel is filled with 2.06 g (0.012 mol) of benzyl bromide (Note 13) in 10 mL of tetrahydrofuran (Note 12). Sodium hydride (0.0145 mol, 0.69 g of a 50% dispersion in oil) (Note 14) is added in portions to the flask and, after gentle reflux for 20 min, the mixture is cooled to room temperature and the solution of benzyl bromide is added dropwise via the addition funnel (10–15 min). Tetrabutylammonium iodide (0.48 g, 0.0013 mol) is added in one portion and the solution is stirred overnight at room temperature (Note 15). The solvent is removed under reduced pressure (water aspirator) and the residue is treated with 50 mL of saturated sodium chloride (NaCl) solution. The mixture is extracted with dichloromethane (3 × 50 mL) (dilution of the aqueous phase with water to avoid emulsions may be necessary) and the combined extracts are dried over anhydrous sodium sulfate. The solvent is removed under reduced pressure using a rotatory evaporator and the residue is flash chromatographed (silica, diethyl ether – petroleum ether 1:1) to give 3.53 g (96% yield) of product as an oil (Note 16).
D. Thiazolyl-to-formyl deblocking. The protected 1-(2-thiazolyl)-D-glycitol (3.5 g, 0.0115 mol) is dissolved in 70 mL of acetonitrile (Note 17), methyl iodide (24.7 g, 0.173 mol, purified by passing through neutral alumina) is added and the resulting mixture is heated to reflux until N-methylation of thiazole is complete (at least 24 hr) as shown by TLC (Note 15) and (Note 18). The solvent is removed under reduced pressure and the residue (Note 19) is dissolved in at least 70 mL of methanol. The solution is cooled to 0°C in an ice bath and sodium borohydride (0.65 g, 0.0172 mol) is added in portions while the solution is stirred vigorously. After the solution is stirred for 30 min at 0°C, it is treated with 2 mL of acetone, and the solvent is removed under reduced pressure. The residue is treated with a saturated solution of sodium chloride (50 mL) and extracted with dichloromethane (3 × 50 mL). The combined extracts are dried over anhydrous sodium sulfate and the solvent is removed under reduced pressure. The resulting oil (Note 20) is dissolved in acetonitrile (5 mL) and the solution is slowly added to a vigorously stirred solution of mercury chloride (3.7 g, 0.0136 mol) in a 4:1 mixture of acetonitrile – water (50 mL). After being stirred at room temperature for 15 min, the reaction mixture is filtered on a Büchner funnel through Celite and the sticky inorganic residue is rinsed with diethyl ether. The combined ethereal extracts are added to the filtered solution and the mixture is concentrated to ca. 10 mL by evaporation of the solvent under reduced pressure. The residue is treated with a saturated solution of potassium chloride (KCI) (50 mL), extracted with dichloromethane (3 × 50 mL), and the combined extracts are dried over anhydrous sodium sulfate (Note 21). Removal of volatile material under reduced pressure gives an oil that is flash chromatographed (silica, diethyl ether – petroleum ether 1:1) to afford 1.77 g (62%) of 2-O-benzyl-3,4-isopropylidene-D-erythrose (Note 22).
2. Notes
1.
The glass components of the apparatus were dried overnight in a 150°C-oven and allowed to cool in a
desiccator over a drying agent before assembly.
2.
Diethyl ether was distilled from
sodium wire under a
nitrogen atmosphere immediately prior to use.
3.
A 1.5 M solution of
butyllithium in hexane was purchased from Aldrich Chemical Company, Inc. Aliquots were transferred to a
100-mL graduated cylinder, stoppered with a
rubber septum using a
15-gauge cannula and
argon.
4.
The slight excess of
butyllithium with respect to
chlorotrimethylsilane is used to avoid acid-catalyzed protodesilylation of 2-TST during the work-up operations.
5.
2-Bromothiazole, available from Aldrich Chemical Company, Inc., was distilled prior to use.
6.
Chlorotrimethylsilane was obtained from Fluka Chemical Corporation and distilled before use.
7.
The Claisen distillation head was filled with glass wool to avoid formation of foam. The checkers found that constant heating of the distillation apparatus with a heat gun greatly facilitates the rate of distillation and minimizes the column holdup.
8.
The product showed the following spectroscopic properties:
1H NMR (80 MHz, CDCl
3, TMS) δ: 0.40 (s, 9 H), 7.40 (d, 1 H, J = 3.0), 8.01 (d, 1 H, J = 3.0);
13C NMR (75.5 MHz, CDCl
3, TMS) δ: −1.20 (q), 121.3 (d), 145.8 (d), 174.3 (s). Other physical properties includes n
22.5D 1.4975 and d = 0.987.
9.
The aldehyde was freshly distilled material prepared according to the
Organic Syntheses procedure.
210.
Dichloromethane was freshly distilled under a
nitrogen atmosphere from
calcium hydride.
11.
This product (anti-adduct) was ≥95% diastereomerically pure based on comparison of the
1H NMR spectrum with that of the syn-adduct.
3 Physical properties and spectral data are as follows:
[α]D −1.2° to −1.5° (MeOH,
c 1.0)
1H NMR (80 MHz, CDCl
3-D
2O, TMS) δ: 1.40 (s, 3 H), 1.47 (s, 3 H), 4.0 (m, 2 H), 4.45 (m, 1 H), 5.07 (d, 1 H, J = 5.1), 7.30 (d, 1 H, J = 3.2), 7.73 (d, 1 H, J = 3.2);
13C NMR (75.5 MHz, CDCl
3, TMS) δ: 25.16 (q), 26.69 (q), 65.38 (t), 71.77 (d), 78.42 (d), 110.54 (s), 120.33 (d), 142.96 (d), 170.95 (s).
12.
Tetrahydrofuran was distilled from
lithium aluminum hydride under a
nitrogen atmosphere immediately prior to use.
13.
Benzyl bromide was purchased from Aldrich Chemical Company, Inc., and purified by passing through neutral alumina.
14.
Sodium hydride, 60% dispersion in mineral oil from Aldrich Chemical Company, Inc., was used as obtained.
15.
The reaction appeared complete by TLC (silica,
diethyl ether –
petroleum ether, 1:1); R
f alcohol = 0.15, R
f O-benzyl derivative = 0.27.
16.
Physical properties and spectral data are as follows:
[α]D +53.7° to +59.6° (CHCl
3,
c 1.66);
1H NMR (270 MHz, CDCl
3, TMS) δ: 1.34 (s, 3 H), 1.39 (s, 3 H), 4.02 (dd, 1 H, J = 4.9, 1.2), 4.52–4.70 (m, 3 H), 4.83 (d, 1 H, J = 5.5), 7.33–7.35 (m, 5 H), 7.39 (d, 1 H, J = 3.2), 7.81 (d, 1 H, J = 3.2);
13C NMR (75.5 MHz, CDCl
3, TMS) δ: 25.39 (q), 26.54 (q), 56.31 (t), 72.81 (t), 78.65 (d), 79.09 (d), 110.67 (s), 120.73 (d), 128.74 (d), 128.86 (d), 129.20 (d), 138.0 (s), 143.5 (s) 170.55 (s).
17.
Reagent grade acetonitrile from Carlo Erba was used as obtained.
18.
The R
f of the
N-methylthiazolium salt is zero.
19.
A sample of this material obtained in a separate experiment was crystallized from
methanol –
diethyl ether to give the pure
N-methylthiazolium iodide as white crystals, mp
181–183°C (dec);
1H NMR (80 MHz, CD
3OD, TMS) δ: 1.33 (s, 3 H), 1.51 (s, 3 H), 3.95–4.51 (m, 3 H), 4.17 (s, 3 H), 4.79 (br d, 2 H), 5.36 (d, 1 H, J = 7.2), 7.34 (s, 5 H), 8.27 (m, 2 H).
20.
A sample of this material obtained in a separate experiment was purified by chromatography (silica,
dichloromethane –
diethyl ether 85:5) to give the pure
thiazolidine as 1:1 mixture of diastereoisomers : oil;
1H NMR (80 MHz, CDCl
3, TMS) δ: 1.36 (s, 3 H), 1.42 (s, 3 H), 2.31 (s, 1.5 H), 2.33 (s, 1.5 H), 2.77–3.25 (m, 4 H), 3.66 (m, 1 H), 3.90–4.55 (m, 4 H), 4.77 (s, 2 H), 7.32 (s, 5 H).
21.
The solid residue contains mercury salts and should be disposed of by procedures used for heavy metal residues.
22.
The product showed the following properties: oil;
[α]D +36.8° to +37.1° (CHCl
3,
c 1.70); IR (film) cm
−1: 2720, 1734;
1H NMR (270 MHz, CDCl
3, TMS) δ: 1.35 (s, 3 H), 1.43 (s, 3 H), 3.81 (dd, 1 H, J = 6.8, 2.5), 3.92 (dd, 1 H, J = 6.1, 9.6), 4.07 (dd, 1 H, J = 9.2, 7.3), 4.35 (m, 1 H), 4.67 (AB quartet, 2 H, J = 11.6), 7.3–7.38 (m, 5 H), 9.70 (d, 1 H, J = 2.5);
13C NMR (75.5 MHz, CDCl
3, TMS) δ: 25.22 (q), 26.55 (q), 66.37 (t), 73.51 (t), 75.22 (d), 83.31 (dd), 110.24 (s), 128.38 (d), 128.75 (d), 137.23 (s), 201.4 (d).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
2-(Trimethylsilyl)thiazole (2-TST)4 is used as a stable and convenient substitute for
2-lithiothiazole for introducing various substituents at C-2 of the thiazole ring (
Figure 1). Reactions of 2-TST with various carbon electrophiles
5 occur readily under mild conditions without the need for any catalyst to give the corresponding 2-substituted thiazoles in fair yields. As has been already pointed out,
5 the multigram preparation of 2-TST from
2-bromothiazole via halogen-metal exchange is much more convenient and practical than the procedure
6 employing the highly volatile and expensive unsubstituted
thiazole.
Figure 1. Synthesis of 2-Substituted Thiazoles from 2-TST
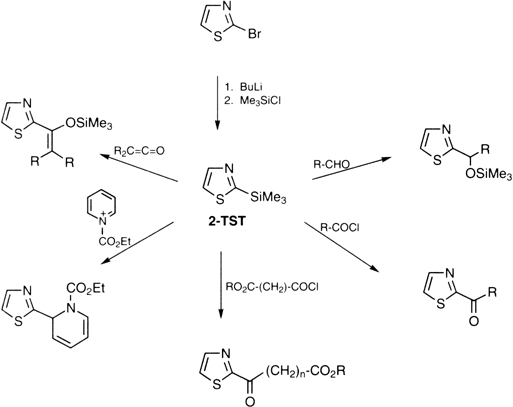
The procedure presented here illustrates the use of 2-TST as a one-carbon homologating reagent of a chiral α,β-dialkoxy aldehyde.
3,7 The protocol is based on three essential key operations, i.e., the anti-stereoselective addition (ds ≥ 95%) of 2-TST to the aldehyde (Step B), the protection of the
hydroxy group at the newly formed stereogenic center (Step C), and the liberation of the formyl group from the thiazole ring (Step D). This step involves a sequence of three reactions (N-methylation of the thiazole ring, reduction, and hydrolysis) that occurs under almost neutral conditions and leaves unaltered the asymmetric centers in the chiral compounds. The procedure for the N-methylation appears more practical than an earlier method
8 employing
trimethyloxonium fluoroborate in liquid
sulfur dioxide.
9 Overall, 2-TST appears to serve as an equivalent to the formyl anion synthon. The iterative application of this principle over several consecutive cycles produces a series of homologues of
D-glyceraldehyde up to a nine-carbon chain and with an all-
anti configuration of the vicinal hydroxy groups.
3 This linear iterative one-carbon extension technology was successfully applied to
L-threose acetonide,
3 dialdoses,
3,10 and α-amino aldehydes.
11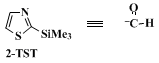
Although various compounds are known to act as synthetic equivalents to the formyl anion synthon,
12 the advantages that appear to be associated with the use of 2-TST warrant comment. First, 2-TST is a readily available, relatively cheap, and storable reagent; second, it reacts promptly and stereoselectively with aldehydes under neutral conditions; third, it gives high yields of products that are stable to isolation and purification and nevertheless can be readily transformed into aldehydes.
This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
petroleum ether
D-glyceraldehyde acetonide
(1R)-2,3-Di-O-isopropylidene-1-(2-thiazolyl)-D-glycitol
(1R)-O-Benzyl-2,3-di-O-isopropylidene-1-(2-thiazolyl)-D-glycitol
2-O-Benzyl-3,4-isopropylidene-D-erythrose
D-(R)-GLYCERALDEHYDE ACETONIDE
1,3-Dioxolane-4-acetaldehyde, 2,2-dimethyl- α-(phenylmethoxy)-, [R-(R*,R*)]-
2-(Trimethylsilyl)thiazole (2-TST)
Thiazolyl-to-formyl deblocking
1-(2-thiazolyl)-D-glycitol
L-threose acetonide
methanol (67-56-1)
diethyl ether (60-29-7)
acetonitrile (75-05-8)
sodium bicarbonate (144-55-8)
sodium chloride (7647-14-5)
sulfur dioxide (7446-09-5)
bromobutane (109-65-9)
sodium sulfate (7757-82-6)
nitrogen (7727-37-9)
acetone (67-64-1)
sodium (13966-32-0)
hydroxy (3352-57-6)
mercury chloride (7487-94-7)
Methyl iodide (74-88-4)
dichloromethane (75-09-2)
potassium chloride (7447-40-7)
D-glyceraldehyde (56-82-6)
benzyl bromide (100-39-0)
butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
lithium aluminum hydride (16853-85-3)
thiazole (288-47-1)
sodium hydride (7646-69-7)
hexane (110-54-3)
argon (7440-37-1)
calcium hydride (7789-78-8)
sodium borohydride (16940-66-2)
trimethyloxonium fluoroborate (420-37-1)
CHLOROTRIMETHYLSILANE (75-77-4)
Tetrabutylammonium fluoride (429-41-4)
tetrabutylammonium iodide (311-28-4)
2-(Trimethylsilyl)thiazole (79265-30-8)
2-bromothiazole (3034-53-5)
N-methylthiazolium
N-methylthiazolium iodide
thiazolidine (504-78-9)
2-lithiothiazole
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved