Org. Synth. 2012, 89, 323-333
DOI: 10.15227/orgsyn.089.0323
Discussion Addendum for:
[Diastereoselective Homologation of D-(R)-Glyceraldehyde Acetonide Using 2-(Trimethylsilyl)thiazole: 2-O-Benzyl-3,4-isopropylidene-D-erythrose]
Submitted by Alessandro Dondoni
*1 and Alberto Marra.
Discussion
Introduction
Over the last several decades, numerous heterocycles have emerged as useful tools (precursors, reagents, chiral auxiliaries) in new methodologies for organic synthesis.
2 In that context, the five-membered heterocycle
1,3-thiazole attained considerable popularity by virtue of its stability under a wide range of reaction conditions, as well as its facile conversion into the formyl group by a very efficient three-step one-pot procedure.
3 The widespread use of the thiazole ring as masked formyl group led to the formulation of a general synthetic strategy that we have referred to as the "Thiazole-Aldehyde Synthesis".
4 Thus, the stereoselective synthesis of a chiral α-hydroxy aldehyde (
aldehydo-D-erythrose) by one-carbon chain homologation of D-glyceraldehyde using
2-trimethylsilylthiazole (2-TST or TMST) as a formyl anion equivalent (see scheme above) represented the first example of diastereoselective thiazole-based synthesis reported from our laboratory in 1986.
Notably, this reaction sequence was repeated over several consecutive cycles, with the result being that various chiral polyhydroxylated aldehydes (
aldehydo sugars) with up to eight carbon atoms were prepared.
5 Moreover, this strategy was applied also to dialdoses to give, via elongation of the side chain, homologues with up to nine carbon atoms.
6 Thus, as 2-TST can be easily prepared by an efficient procedure (see the original paper in this journal), or can be purchased at reasonable price from a number of suppliers, its use in the Thiazole-Aldehyde Synthesis over the past 25 years became routine.
Most notable is the fact that 2-TST reacts readily with aldehydes and other
C-electrophiles (acyl chlorides, pyridinium chlorides, ketenes),
7 as well as sulfenyl halides,
8 without the need of any catalytic fluoride ion.
These fluoride-free
C- and
S-desilylation processes had no precedents in silicon chemistry as they proceed by a special mechanism involving a thiazolium ylide intermediate.
9Scope of the 2-TST-Based Thiazole-Aldehyde Synthesis
The key role of 2-TST (often referred to as the
Dondoni reagent) serving as a masked formyl anion equivalent has been reviewed in a comprehensive article
3 covering the period ranging from its first synthesis
10 in 1981 up to 2003.
Nevertheless, some examples highlighting the role of 2-TST in synthetic routes leading to biologically active compounds are reported again below in Schemes 1-10 of this discussion addendum.
Scheme 1. Synthesis of the disaccharide subunit of bleomycin A
2 from
aldehydo-L-xylose.
11
Scheme 2. Synthesis of the oligosaccharide portion of everninomicin 13,384-1 from
aldehydo-L-threose.
12
Scheme 3. Synthesis of an acetylated phytosphingosine from
N-Boc serinal acetonide.
13
Scheme 4. Synthesis of a protected amino diol, a building block of renin inhibitors, from
N-Boc cyclohexylalaninal.
14
Scheme 5. Synthesis of a hydroxyethylamine isosteric dipeptide precursor of Saquinavir (Ro 31-8959) from
N-Boc phenylalaninal (Hoffman-La Roche method).
15
Scheme 6. Synthesis of a hydroxyethylamine isosteric dipeptide precursor of Saquinavir (Ro 31-8959) from
N-Boc,
N-PMB phenylalaninal (Dondoni method).
16
Scheme 7. Synthesis of a chiral pseudo
C2-symmetric 1,3-diamino-2-propanol derivative from
N-Boc phenylalaninal.
17
Scheme 8. Synthesis of
N-benzoyl 3-phenylisoserine, viz the side chain of Taxol, from
N-benzoyl phenylglycinal.
18
Scheme 9. Synthesis of
N-Boc and acetonide protected 5-
O-carbamoyl polyoxamic acid from chiral epoxy butanal.
19
Scheme 10. Synthesis of the amino acid side chain of the pseudopeptide microbial agent AI-77-B from a formyl oxazolidine.
20
Even with the development of new research methods, the Thiazole-Aldehyde Synthesis based on the use of 2-TST continued to be an attractive synthetic tool.
Thus, recent applications of this chemistry that have been carried out in our own and other laboratories are reported below.
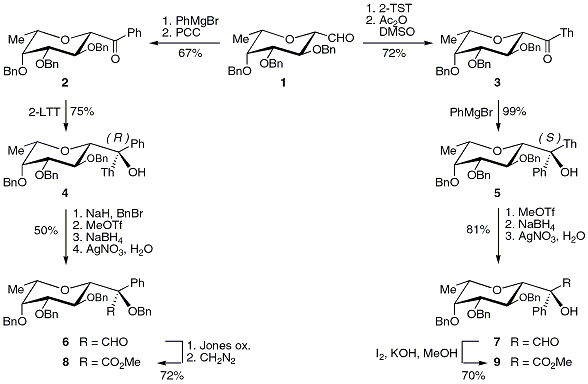
In the context of a study on the synthesis of
C-fucosides of biological relevance, we set out to develop the syntheses of
R- and
S-epimeric
C-fucosyl hydroxyphenyl acetates
21 (Scheme 11).
These glycoconjugates were expected to constitute the glycosidic part of new antibiotics similar to existing natural products.
22 The β-L-
C-fucosyl aldehyde
1, which was prepared in our laboratory, was utilized as the starting material for the synthesis of both epimers.
The chiral alcohols
4 and
5 with
R and
S configuration at quaternary carbon atoms, respectively, were produced by using essentially the same reagents, but employing them in an inverse order.
For example, aldehyde
1 was first reacted with phenylmagnesium bromide (PhMgBr) and the resulting mixture of diastereomeric alcohols was oxidized to give the ketone
2. Addition of
2-lithiothiazole (2-LTT) to this ketone afforded the
R-configured tertiary alcohol
4. For the preparation of the alternate epimer, the sequence was initiated by the addition of 2-TST to the aldehyde
1 and oxidation of the mixture of epimeric alcohols to ketone
3 followed by addition of PhMgBr to furnish the
S-configured alcohol
5. Having both stereoisomeric tertiary alcohols
4 and
5 in hand, their absolute configuration was assigned from their
1H NMR spectra (NOE experiments).
Finally, both thiazolyl alcohols were readily transformed in the corresponding aldehydes
6 and
7 by the standard thiazole-to-formyl unmasking protocol and these aldehydes were oxidized to the representative esters
8 and
9.
Scheme 11. Syntheses of R- and S-epimer C-fucosyl hydroxyphenyl acetaldehydes and acetates.
Another paper on the use of 2-TST in the Thiazole-Aldehyde Synthesis that appeared in mid-2005 was reported by Alcaide, Almendros, and their co-workers.
23 These authors developed a synthetic route leading to
azabicyclo[4.3.0]nonane (indolidizinone) amino esters from β-lactams (Scheme 12) by using α-alkoxy β-lactam acetaldehydes as key intermediates.
Interest in the synthesis of indolizidinone amino acids stemmed from their behavior as conformationally restricted dipeptide mimetics.
Thus, the readily available enantiopure 4-oxoazetidine-2-carbaldehydes
10 and
11 were transformed into the one-carbon higher homologues via stereoselective addition of 2-TST to give the thiazole derivatives
12-
13 and unmasking of the formyl group from the thiazole ring (Scheme 12).
In all cases, well established conditions
4 for 2-TST addition to aldehydes, as well as for thiazole cleavage, were employed resulting in the isolation of the α-alkoxy β-lactam acetaldehydes
14 and
15 in good yields.
These aldehydes were transformed into the target indolizidinone amino esters
16 and
17 via standard heterocyclic chemistry.
A similar 2-TST-based strategy was followed by the same research group for the conversion of enantiopure
cis-4-formyl-1-(3-methyl-2-butenyl)-3-methoxy-β-lactam into the corresponding higher homologue, an α-silyloxylated aldehyde that served as key intermediate for the synthesis of a mixture of diastereomeric dihydroxycarbacephams via an intramolecular carbonyl-ene reaction.
24 It was emphasized in both instances that the addition of 2-TST to 4-oxoazetidine 2-carbaldehydes to give the α-hydroxyalkylthiazoles was not a trivial process as the same authors had previously observed the reaction of 2-TST with
N-aryl-4-formyl-β-lactams to give enantiopure α-alkoxy-γ-keto acid derivatives via N1-C4 bond cleavage of the β-lactam ring.
25Scheme 12. Synthesis of α-alkoxy β-lactam acetaldehydes from 4-oxoazetidine-2-carbaldehydes en route to azabicyclo[4.3.0]nonane (indolidizinone) amino esters.
![Scheme 12. Synthesis of α-alkoxy β-lactam acetaldehydes from 4-oxoazetidine-2-carbaldehydes en route to azabicyclo[4.3.0]nonane (indolidizinone) amino esters.](/content/figures/V89P0323-13.gif)
A fourth paper on the use of 2-TST was reported in 2007 by Cateni and co-workers in the effort to prepare a natural cerebroside from Euphorbiaceae.
26 These authors took advantage of the availability of the
O- and
N-protected amino sugar
18 (L-
ribo configuration) that was previously prepared in our laboratory by 2-TST-based homologation of the Garner aldehyde.
13 Thus, the
aldehydo sugar
18 was transformed into the one-carbon higher homologue
19 by the standard 2-TST-based methodology (Scheme 13).
Wittig reaction of this
aldehydo sugar (L-
allo configuration) with a long chain alkyl phosphorane afforded under suitable conditions the
Z-olefin
20 displaying a chiral polar head and a lipophilic tail.
Compound
20 was, in fact, the required C
18-sphingosine, which after glycosylation and acylation at the polar head, led to the targeted natural cerebroside.
Scheme 13. Synthesis of a C18-sphingosine intermediate in the synthesis of a natural cerebroside.
Conclusion
From the selected examples reported in the above schemes, it appears that since its discovery in 1981, 2-TST has found use as a formyl anion equivalent in various synthetic routes leading to compounds of biological relevance.
The fidelity of this reagent to react
spontaneously with aldehydes bearing a wide variety of substituents has been amply demonstrated.
It has to be mentioned, however, that the synthetic utility of 2-TST extends beyond its use in the Thiazole-Aldehyde Synthesis, as in recent years numerous applications of this reagent have appeared in thiazole chemistry
27 and in synthetic routes leading to thiazole-containing compounds of biological relevance.
28
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2-O-Benzyl-3,4-isopropylidene-D-erythrose
1,3-thiazole (288-47-1)
2-(Trimethylsilyl)thiazole,
(79265-30-8)
2-lithiothiazole
D-(R)-GLYCERALDEHYDE ACETONIDE (15186-48-8)
 |
Alessandro Dondoni was born and grew up in Ostiglia, Italy.
He received a degree (laurea) in industrial chemistry from the University of Bologna in 1960.
After postdoctoral studies at the IIT in Chicago with Sidney I.
Miller, he returned to the University of Bologna in 1963 where he began his independent scientific career.
In 1973 he joined the University of Ferrara and was named Professor of organic chemistry in 1975.
He retired in 2008 and at present (2012) is Senior Research Scientist in the same University.
At the beginning of his career he was involved in studies of the kinetics and mechanism of 1,3-dipolar cycloaddition reactions and physicochemical properties of sulfur compounds, while more recently he has focused on the development of new synthetic methods for heterocyclic and carbohydrate chemistry.
|
 |
Alberto Marra graduated in Pharmaceutical Sciences from the University of Pisa (Italy) and obtained his Ph.D.
degree in Organic Chemistry from the University Paris VI (France) under the supervision of Prof.
P.
Sinay.
After a postdoctoral fellowship at the University of Zurich (Switzerland) with Prof.
A.
Vasella, he was appointed to a lectureship in Organic Chemistry at the University of Ferrara, where he joined the group of Prof.
A.
Dondoni.
In 1998 he was promoted to the position of Associate Professor at the same university.
He was Visiting Professor at the Université de Picardie (2008) and the Université de Cergy-Pontoise (2010). |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved