Org. Synth. 1997, 74, 194
DOI: 10.15227/orgsyn.074.0194
A SIMPLE AND CONVENIENT METHOD FOR THE PREPARATION OF (Z)-β-IODOACROLEIN AND OF (Z)- OR (E)-γ-IODO ALLYLIC ALCOHOLS: (Z)- AND (E)-1-IODOHEPT-1-EN-3-OL
[2-Propenal, 3-iodo-, (Z)- and 1-Hepten-3-ol, 1-iodo-, (Z)- and (E)-]
Submitted by Ilane Marek, Christophe Meyer, and Jean-F. Normant
1.
Checked by David A. Favor and Amos B. Smith, III.
1. Procedure
A. Ethyl (Z)-β-iodoacrylate (Note 1). A 250-mL, round-bottomed flask equipped with a magnetic stirring bar and nitrogen gas inlet is charged with 30.6 g (204 mmol) of dry sodium iodide (Note 2) and 100 mL of glacial acetic acid. To the stirred solution is added in one portion 20.6 mL (204 mmol) of ethyl propiolate (Note 2) and the resulting mixture is heated with an oil bath (bath temperature 70°C) for 12 hr. The brown solution is cooled to room temperature, and 100 mL of water and 100 mL of ether are added. The organic layer is separated and the aqueous layer is extracted with ether (2 × 20 mL). The combined organic layers are treated with 3 M aqueous potassium hydroxide (ca. 150 mL in 50-mL portions) until the aqueous phase becomes neutral (pH = 7), washed with 50 mL of brine, and dried over anhydrous magnesium sulfate. After evaporation of the solvent, the residual brown oil is distilled to give 40.6 g (88% yield) of ethyl (Z)-β-iodoacrylate (Note 3) as a pale yellow liquid, bp 57°C/0.1 mm.
B.
(Z)-1-Iodohept-1-en-3-ol. A 100-mL, dry
(Note 4),
four-necked, round-bottomed flask equipped with a mechanical stirrer, an internal thermometer, a rubber septum, and a nitrogen gas inlet, is charged with
2.26 g (10 mmol) of ethyl (Z)-β-iodoacrylate, 2, and
20 mL of anhydrous dichloromethane (Note 5). The stirred solution is cooled to −78°C by means of a
liquid nitrogen bath and
10 mL (10 mmol) of a 1 M solution of diisobutylaluminum hydride in hexane (Note 6) is added dropwise with a syringe at such a rate that the temperature does not exceed −75°C. After stirring for 30 min at −78°C
(Note 7) 11 mL (11 mmol, 1 M solution in Et2O) of a butylmagnesium bromide solution in
ether (Note 8) is added dropwise at −70°C with a syringe through the septum. The cooling bath is removed and the reaction mixture is allowed to warm to room temperature. Hydrolysis is carried out at −20°C by dropwise addition of
20 mL of a 1 M aqueous solution of hydrochloric acid, followed by addition of
30 mL of ether. The organic layer is separated, the aqueous layer is extracted with
ether (2 × 20 mL), and the combined extracts are dried over
magnesium sulfate. After rotary evaporation of the solvents, the residual pale yellow oil is purified by chromatography through
1.85 g of silica (Note 9) packed in a
4-cm diameter column and eluted with
15% ethyl acetate in hexane, to give
1.7 g of
(Z)-1-iodohept-1-en-3-ol,
2, (
71% yield) as a pale yellow liquid
(Note 10).
C.
(E)-1-Iodohept-1-en-3-ol. A 100-mL, dry
(Note 4) four-necked, round-bottomed flask equipped with a mechanical stirrer, an internal thermometer, a rubber septum, and a nitrogen gas inlet, is charged with
2.26 g (10 mmol) of ethyl Z-β-iodoacrylate, 2, and
20 mL of anhydrous dichloromethane (Note 5). The stirred solution is cooled to −78°C by means of a liquid nitrogen bath and
10 mL (10 mmol) of a 1 M solution of diisobutylaluminum hydride in hexane (Note 6) is added dropwise with a syringe at such a rate that the temperature does not exceed −75°C. After the mixture is stirred for 30 min at −78°C
(Note 7), it is allowed to warm slowly to 0°C in 45 min, stirred for 15 min at this temperature, and then cooled to −20°C;
11 mL (11 mmol, 1 M solution in Et2O) of a butylmagnesium bromide solution in ether (Note 8) is added dropwise at −20°C with a syringe through the septum. The reaction mixture is allowed to warm to room temperature and worked up, as described for the Z isomer.
(E)-1-Iodohept-1-en-3-ol (
1.9 g) is obtained as a colorless liquid (
79% yield)
(Note 11) with an E/Z ratio of 96/4 .
D.
(Z)-β-Iodoacrolein, 4. A 100-mL, dry
(Note 4),
four-necked, round-bottomed flask equipped with a mechanical stirrer, an internal thermometer, a rubber septum, and a nitrogen gas inlet, is charged with
2.26 g (10 mmol) of ethyl (Z)-β-iodoacrylate, 2, and
20 mL of anhydrous dichloromethane (Note 5). The stirred solution is cooled to −78°C in a liquid nitrogen bath and
10 mL (10 mmol) of a 1 M solution of diisobutylaluminum hydride in hexane (Note 6) is added dropwise with a syringe at such a rate that the temperature does not exceed −75°C. After the solution is stirred at −78°C
(Note 7),
5 mL of cold methanol (Note 12) is added dropwise through the septum with a syringe at −80°C. Immediately after this addition,
25 mL of aqueous 20% potassium sodium tartrate is added to the cold reaction mixture in one portion, which causes the temperature to reach 0°C within a few seconds. The cooling bath is removed and
20 mL of ether is added to the hydrolyzed reaction mixture, which is further stirred for 20 min at room temperature
(Note 13). It is then filtered through a pad of Celite and extracted with
30 mL of ether. The organic layer is washed with
brine and dried over
potassium carbonate. After careful rotary evaporation of the solvents,
1.6 g of
(Z)-β-iodoacrolein is obtained as a yellow liquid (
88%)
(Note 14), which can be handled easily as
ether or
dichloromethane solutions, and can be stored in the refrigerator for a few weeks without decomposition or isomerization.
2. Notes
1.
Part A of the procedure should be carried out in an
efficient fume hood to avoid exposure to noxious vapor (
acetic acid) and to lachrymatory
ethyl propiolate.
2.
Sodium iodide, 99%, was purchased from Janssen Chimica and used as received.
Glacial acetic acid, 99%, was purchased from Prolabo.
Ethyl propiolate was obtained from Janssen Chimica and used as received.
3.
The product exhibits the following physical and spectral properties: IR (film) cm
−1: 3080, 2940, 1735, 1640, 1425, 1365, 1230, 1170, 1020, 925, 730;
1H NMR (400 MHz, CDCl
3) δ: 1.32 (t, 3 H, J = 7.15, C
H3CH
2), 4.25 (q, 2 H, J = 7.14, OC
H2), 6.89 (d, 1 H, J = 9.34, =C
H-CO
2Et), 7.44 (d, 1 H, J = 9.34, =C
HI);
13C NMR (100 MHz, CDCl
3) δ: 14.2, 60.8, 94.6, 129.9, 164.6.
4.
All glassware is oven dried at 140°C overnight and assembled while hot under a
nitrogen atmosphere.
5.
Dichloromethane was distilled from
calcium hydride and stored over 4 Å molecular sieves.
6.
Diisobutylaluminum hydride was purchased from Aldrich Chemical Company, Inc.7.
At this point, TLC analysis of a hydrolyzed (1 M aqueous
hydrochloric acid) aliquot, eluted with
10% ethyl acetate in hexane, indicated that the reaction is complete.
8.
Butylmagnesium bromide was prepared from the corresponding
butyl bromide and
magnesium turnings in
anhydrous ether.
29.
Silica gel was purchased from Merck: Geduran SI 60.
10.
The product exhibits the following spectral properties:
1H NMR (400 MHz, CDCl
3) δ: 0.90 (t, 3 H, J = 7.15, C
H3), 1.31–1.36 (m, 4 H), 1.51–1.63 (m, 2 H), 1.7 (s, 1 H, O
H), 4.4 (m, 1 H, C
HOH), 6.20 (t, 1 H, J = 7.69, =C
H), 6.33 (d, 1 H, J = 7.69, =C
HI);
13C NMR (100 MHz, CDCl
3) δ: 14.0, 22.6, 27.1, 35.6, 74.4 (CHOH), 82.2 (=CI), 143.4 (=CH).
11.
The product exhibits the following spectral properties: IR (film) cm
−1: 3350, 3060, 2950, 2920, 2850, 1605, 1450, 1265, 1165, 1015, 930;
1H NMR (400 MHz, CDCl
3) δ: 0.90 (t, 3 H, J = 7.15, C
H3), 1.31–1.36 (m, 4 H), 1.51–1.63 (m, 2 H), 1.71 (s, 1 H), 4.1 (m, 1 H, C
HOH), 6.34 (dd, 1 H, J = 14.3, 1.1, =C
HI), 6.58 (dd, 1 H, J = 14.3, 6.6, =C
H);
13C NMR (100 MHz, CDCl
3) δ: 13.9, 22.5, 27.2, 36.2, 74.6 (CHOH), 77.2 (=CI), 148.6 (=CH).
12.
Methanol was obtained from Merck & Company, Inc., and used as received.
13.
The workup should be carried out under an efficient fume hood since the title compound is a lachrymator.
14.
The
(Z)-β-iodoacrolein exhibits the following spectral properties: IR (CCl
4) cm
−1: 3050, 2820, 2730, 1675, 1610, 690;
1H NMR (400 MHz, CDCl
3) δ: 6.77 (dd, 1 H, J = 8.25, 6.6, =C
H-CHO), 7.79 (d, 1 H, J = 8.25, =C
HI), 9.67 (d, 1 H, J = 6.6, C
HO);
13C NMR (100 MHz, CDCl
3) δ: 103.0 (=CHI), 136.4 (=CH), 195.4 (C=O).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The present procedure illustrates the simplest convenient method for the preparation of (Z)- or (E)-γ-iodo allylic alcohols and
(Z)-β-iodoacrolein. It uses
ethyl propiolate as a unique starting material:
ethyl propiolate and
ethyl tetrolate can be hydroiodinated regio- and stereospecifically (Z > 99%) by reaction with inexpensive
sodium iodide in
acetic acid3 4 5 6 7 8 (Scheme 1). The same methodology has been applied recently to the synthesis of enantiomerically pure (Z)-2-haloalkenyl sulfoxides.
9 By reaction of
2 with
diisobutylaluminum hydride at low temperature, and subsequent reaction with a Grignard reagent, one can obtain, very easily, the secondary (Z)-γ-iodo allylic alcohols with good chemical yield and exclusive Z stereochemistry of the double bonds (Table I). This one-pot "reduction-C-alkylation" sequence of the ester group allows the preparation of very sensitive derivatives (entry 5 or 6), which are difficult to prepare by usual methods.
10 11 12 13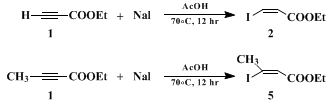
TABLE I
|
|
Starting Ester
|
Grignard Reagents
|
Products
|
Yield
|
|
|
|
EtMgBr
|
|
88%
|
|
|
BuMgBr
|
|
79%
|
|
|
PhMgBr
|
|
85%
|
|
|
|
|
72%
|
|
|
|
|
81%
|
|
|
|
|
63%
|
|
|
|
|
65%
|
|
|
BuMgBr
|
|
81%
|
|
However, reaction of
ethyl (Z)-β-iodoacrylate with DIBAL-H at low temperature (−78°C, 15 min) followed by warming to 0°C and addition of a Grignard reagent (−20°C to 0°C) now leads to the E isomers of the secondary
γ-iodo allylic alcohols with an E/Z ratio of 96/4 (Table II). This isomerization can be rationalized as occurring by the influence of the weak Lewis acid, Al(iBu)
2OEt, generated by α-elimination of the thermally labile aluminooxyacetal.
14 The
(Z)-β-iodoacrolein 4 and
crotonaldehyde 6 (Scheme 2) are also easily obtained by dropwise addition of an excess of
methanol to the aluminooxyacetal at low temperature, immediately followed by alkaline hydrolysis. (3Z)-Iodopropenals are valuable intermediates in organic synthesis because of the presence of three functional groups on three
carbon atoms.
14,15
TABLE II
|
|
Starting Ester
|
Grignard Reagents
|
Products
|
Yield
|
|
|
|
|
|
80%
|
|
|
BuMgBr
|
|
86%
|
|
|
|
|
72%
|
|
Starting from ethyl (Z)-β-iodoacrylate, (E)- or (Z)-γ-iodo allylic alcohols are easily obtained in a highly stereoselective manner by a "reduction-C-alkylation" of the ester group without alteration of the nature or geometry of the iodo vinylic part. (Z)-β-Iodoacrolein can be easily isolated in good yields, and gives rise to various derivatives.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
silica gel
silica
brine
1-Hepten-3-ol, 1-iodo-, (Z)- and (E)-
(Z)-ß-bromoacrolein
(Z)-ß-chloroacrolein
potassium carbonate (584-08-7)
hydrochloric acid (7647-01-0)
acetic acid (64-19-7)
ethyl acetate (141-78-6)
methanol (67-56-1)
ether (60-29-7)
magnesium (7439-95-4)
Butyl bromide (109-65-9)
nitrogen (7727-37-9)
carbon (7782-42-5)
potassium hydroxide (1310-58-3)
sodium iodide (7681-82-5)
Butylmagnesium bromide (693-03-8)
dichloromethane (75-09-2)
magnesium sulfate (7487-88-9)
hexane (110-54-3)
crotonaldehyde (123-73-9)
calcium hydride (7789-78-8)
diisobutylaluminum hydride (1191-15-7)
ethyl propiolate (623-47-2)
Ethyl tetrolate (4341-76-8)
(E)-1-Iodohept-1-en-3-ol (151160-08-6)
(Z)-β-IODOACROLEIN,
2-Propenal, 3-iodo-, (Z)- (138102-13-3)
(Z)-1-Iodohept-1-en-3-ol (138102-06-4)
potassium sodium tartrate (304-59-6)
Ethyl (Z)-β-iodoacrylate,
ethyl Z-β-iodoacrylate (31930-36-6)
β-Iodoacrolein
γ-iodo allylic alcohols
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved