Org. Synth. 2025, 102, 534-561
DOI: 10.15227/orgsyn.102.0534
Synthesis of 4,4-Dimethyl-1,6-Heptadiyne and Derivatives from Dimedone
Submitted by Amir Tavakoli, Nathaniel J. Selvaraj, and Gregory B. Dudley
*1Checked by Aleksa Milosavljevic and Dirk Trauner
1. Procedure (Note 1)
A. Ethyl 5,5-dimethyl-3-oxo-7-octynoate (β-keto ester) (2). 5,5-Dimethyl-3-oxocyclohex-1-en-1-yl trifluoromethanesulfonate (1) was prepared from dimedone (Notes 2 and 3). An oven-dried 250-mL, 1-neck round-bottomed flask A (24/40 joints) was equipped with a 24/40 thermometer adapter (Note 4), glass thermometer (Note 5), a Teflon-coated octagonal stir bar (25 mm x 8 mm), and a rubber septum. The flask was evacuated and purged with argon in triplicate using a syringe needle attached to a Schlenk line (Note 6). Diisopropylamine (9.64 mL, 68.8 mmol, 1.23 equiv) was added to the 250-mL round-bottomed flask A, followed by tetrahydrofuran (THF) (67 mL) (Note 7 and 8) with stirring at room temperature (19-21 ℃) for 5 min at 400 rpm. A separate oven-dried 250-mL round-bottomed flask B (24/40 joint), equipped with an octagonal stir bar (25 mm x 8 mm) and a 24/40 rubber septum, was purged with argon. Diisopropylamine (23.7 mL, 169 mmol, 3.01 equiv) was added to flask B, followed by anhydrous THF (70 mL), with stirring at room temperature (19-21 ℃) for 5 min at 400 rpm. Both flasks were lowered into metal-insulated Dewar flasks. A 1-L, 2-neck (24/40 joints) round-bottomed flask was equipped with an egg-shaped, Teflon-coated stir bar (38 mm x 16 mm) and with a 24/40 thermometer adapter and a glass thermometer (Note 5 and 9). The 1-L round-bottomed flask was purged with nitrogen, placed above the central hot plate, and reserved for later use (Figures 1A-B).

Figure 1. General glassware assembly; A. depiction of Schlenk line, hot plate assembly. Chemglass temperature probe, and glass thermometer employed; B. Enlarged image of diisopropylamine and THF-charged flasks (photos provided by authors)
Separate batches of lithium diisopropylamine (LDA) were prepared as follows. Both round-bottomed flasks A and B were cooled to -78 ℃ using dry ice and acetone and were stirred at this temperature for 10 min at 550 rpm (Note 10, 11, and 12). A solution of n-butyllithium (26.6 mL, 2.5 M in hexane, 67 mmol, 1.2 equiv) was added dropwise (Note 13) to the flask A over 5 min using plastic syringes equipped with oven-dried, 20-gauge, metal needles (Note 14). The solution was stirred at -78 ℃ for 30 min, theoretically producing a 0.64 M solution of LDA. To the flask B, n-butyllithium (66.6 mL, 2.5 M in hexanes, 170 mmol, 3.0 equiv) was added dropwise (using plastic syringes equipped with oven-dried 20-gauge metal needles) (Figure 2). This solution was stirred at -78 ℃ for 30 min, theoretically producing a 1.05 M solution of LDA.
Figure 2. Addition of n-butyllithium via a syringe at -78 ℃ (dry ice/acetone bath) to form LDA solution in THF in flask B (photo provided by checkers)
An oven-dried 100-mL pear-shaped flask (14/20) was equipped with a rubber septum (secured with electric tape) and was purged with argon. The 100 mL flask was charged with dry ethyl acetate (6.50 mL, 66.0 mmol, 1.20 equiv) (Note 15) and anhydrous tetrahydrofuran (45 mL, 1.3 M) at room temperature (19-21 ℃) and the flask was swirled by hand for about 10 seconds for homogeneity. The solution of ethyl acetate in tetrahydrofuran was cannula transferred (20-gauge, oven-dried) into flask A over a 15 min period with stirring at 400 rpm (Note 16 and 17) (Figure 3). After 30 min of stirring at 400 rpm, formation of ethyl lithioacetate was assumed to be complete (TLC A, Note 18).
Figure 3. Cannula transfer of the ethyl acetate solution into the flask containing the LDA solution (photo provided by checkers)
The 1-L round bottom flask that was purged and set aside previously was purged with nitrogen one additional time and charged with the vinylogous acyl triflate (VAT) 1 (15.3 g, 56.7 mmol, 1.0 equiv) and tetrahydrofuran (225 mL, 0.25 M) (Figure 4A). This VAT solution was stirred at 450 rpm at room temperature (19-21 ℃) for 5 min before being transferred to a -78 ℃ dry ice and acetone bath. The yellow VAT solution was cooled to an internal temperature of -68 ℃, at which point the solution of ethyl lithioacetate was transferred from flask A into the 1-L round-bottomed flask over 10 min via a 20-gauge cannula (Note 17). During this addition, the yellow VAT solution became a deep orange color (Figure 4B, Note 19).
Figure 4. General observations for; A. VAT solution at room temperature (19-21 ℃); B. during addition of ethyl lithioacetate (photos provided by authors)
Upon complete addition of ethyl lithioacetate to the solution of VAT 1, stirring proceeded at 450 rpm in the -78 ℃ dry ice and acetone bath for 10 min. The solution was transferred to an ice bath contained in a large glass dish and allowed to warm to -16 ℃, during which time the solution changed from a deep orange to a yellow color (TLC B taken; Figure 7B) (Figure 5A-B).
Figure 5. General observations. A. VAT solution immediately after ethyl lithioacetate addition; B. after 10 min of stirring in an ice bath (photos provided by authors)
After 10 min of stirring in an ice bath, the second solution of lithium diisopropylamine (3.0 equiv) was transferred via a 16-gauge cannula over a 15 min period to the 1-L round-bottomed flask. During this addition of lithium diisopropylamine to the reaction vessel, the color of the solution changed from yellow to a deep red-brown color (Figures 6A-C).
Figure 6: General observations during the addition of 3.00 equiv LDA after A. 5 min; B. 10 min; C. 15 min (photos provided by authors)
Upon complete addition of the additional LDA (3.0 equiv), the reaction vessel was removed from the ice bath and allowed to stir at room temperature (19-21 ℃) for 20 min (Figure 7C). The reaction was quenched with a half-saturated solution of ammonium chloride (400 mL) (Note 20). The solution was transferred to a 1-L separatory funnel with a 35/20 joint, and the aqueous phase and organic phase were separated. The aqueous phase was extracted with ether (50 mL x 4) (Note 21), and the combined organic layers were washed with brine (150 mL x 2) (Note 22). The organic phase was dried with sodium sulfate (Note 23) and vacuum filtered through a 350-mL fritted funnel with a 24/40 adapter attached to a 2-L rotary evaporation flask. The solution was concentrated via rotary evaporation (300-50 mbar) to furnish a crude orange-red oil.
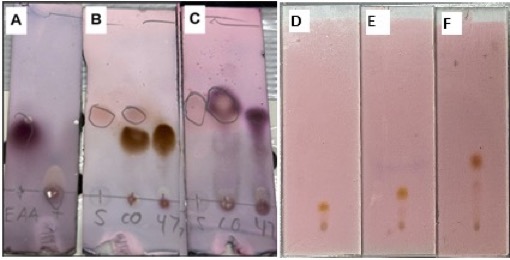
Figure 7. A-C. TLC plates used to monitor the reaction (run in 20 % ethyl acetate/hexanes and visualized with p-anisaldehyde); A. TLC taken of ethyl acetoacetate (EAA; Rf= 0.40 UV(+); pink) and ethyl lithioacetate; B. TLC taken after stirring reaction in ice bath for 10 min with VAT 1 (S; Rf= 0.55 UV (+); light red) co-spotted with the reaction mixture (47; Rf= 0.40 UV(-); orange); C. TLC taken after 3.00 equiv LDA and removing reaction from ice bath with VAT 1 (S) co-spotted against the reaction mixture (47; Rf= 0.50 UV(+); purple) (photos provided by checkers); D-F. TLC plates of the crude reaction mixture after workup, run in; D. 2.5% EtOAc in hexanes; E. 5% EtOAc in hexanes; F. 10% EtOAc in hexanes (photos provided by checkers)
This crude red oil was purified via column chromatography (Notes 24, 25, 26, 27, 28, 29, 30, 31) to furnish β-keto ester 2 (8.56 g, 74 % yield) as an orange oil determined to be of 97% purity via qNMR using 1,3-benzodioxole as internal standard (Note 32). The boiling point of β-keto ester 2 was observed to be 118-119 oC at 13 mbar.
Figure 8. Results of purification by column chromatography; A. color and size of all fractions; B. TLC plates of all fractions (visualized with anisaldehyde stain) (photos provided by authors); C. the column and Erlenmeyer flasks (125-250 mL) used for fraction collection by the checkers; D. the appearance of the final product 2 stored in a 250 mL round-bottomed flask (photo provided by the checkers)
B. 5,5-Dimethyl-octa-2,7-diynoic acid (diyne monoacid, 3). A 100-mL, single neck round bottom flask (14/20 joint), equipped with an octagonal stir bar (25 mm x 8 mm) was charged with lithium hydroxide monohydrate (15.1 g, 359 mmol, 5.0 equiv) and 60 mL of distilled water under open air (Note 33). This affords an approximately saturated solution of lithium hydroxide that was stirred (500 rpm) at room temperature (19-21 ℃) for 15 min before stirring was turned off and residual solid was allowed to settle for 1 h. A separate 500 mL, 3-neck round bottom flask (24/40 joints) was equipped with a 24/40 glass thermometer adapter equipped with a glass thermometer (Note 5 and 9), a 24/40 rubber septum, a 24/40 rubber septum punctured by a venting needle, and an egg-shaped, Teflon-coated stir bar (38 mm x 16 mm) under open air. The 500-mL round-bottomed flask was charged with 8.4 g (40.0 mmol, 1.0 equiv) of β-keto ester 2 followed by 200 mL of hexanes with stirring (500 rpm) at room temperature (19-21 ℃) for 5 min before being transferred to a Dewar filled with acetone and thermostated with an immersion cooler that is set to 2.5 ± 2.0 ℃ (Note 34) (Figures 9A-B). The solution of β-keto ester 2 was cooled to 2.5 ℃, over 15 min, before 40 mL of the lithium hydroxide solution (ca. 5 M) was added in one portion to form a cloudy yellow, biphasic solution (Note 35 and 36). The biphasic solution was stirred (500 rpm) for 10 min, at which point the cloudy yellow solution became a light-yellow emulsion (Note 37).

Figure 9. A. Glassware assembly immersed in a Dewar with acetone thermostated with an immersion cooler; B. immersion cooler set to [2.5 ± 2.0] ℃ (photos provided by checkers); C. syringe pump addition of triflic anhydride to the flask cooled by an ice-water bath (photo provided by authors)
With continued stirring, trifluoromethane sulfonic anhydride (triflic anhydride) (17 mL, 101 mmol, 2.5 equiv) (Note 38) was added via syringe pump over 6 h (Note 39, 40 and 41) (Figure 9C). Upon complete addition of triflic anhydride, the organic and aqueous phases became more homogeneous (Figure 10A) (TLC A taken; Figure 11A). The reaction mixture was stirred for 10 min, and then solid lithium hydroxide monohydrate (17.2 g, 409 mmol, 10.0 equiv) was added slowly over 3 min (Note 42). Tetrahydrofuran (60 mL) was added via syringe pump over 20 min with stirring (TLC B taken; Figure 11B) (Note 43) (Figure 10B). The organic and aqueous phases became indistinguishable (TLC C taken 1 h later; Figure 11C). The reaction mixture was allowed to warm gradually to ambient temperature overnight (18.5 h) with continued stirring (TLC D taken; Figure 11D).
Figure 10: The reaction mixture: (A) after addition of triflic anhydride; and (B) after addition of THF (photos provided by authors)
Figure 11. TLC plates (run in 10 % ethyl acetate/hexanes x 3 and visualized with p-anisaldehyde) used to monitor the reaction (picture taken one day after start of reaction); A. TLC taken after triflic anhydride addition with β-keto ester (S; Rf= 0.70 UV(+); light yellow) co-spotted against the organic phase (67; Rf= 0.75; dark yellow) and aqueous phase (Aq; Rf= 0.00; white); B. TLC taken after addition of THF with (S) co-spotted against the reaction (67; Rf= 0.80; blue); C. TLC taken 1 h later with (S) co-spotted against (67); D. TLC taken 18.5 h after THF addition with (S) co-spotted against (67) (photos provided by authors)
The reaction mixture was transferred to a 1-L separatory funnel, and the organic and aqueous layers were separated (Note 44 and 45). The aqueous phase was extracted with diethyl ether (75 mL x 5), and the organic phases were combined and set aside (Note 46) (Figure 12A). The aqueous phase was transferred to the 1-L separatory funnel and acidified with 6 M HCl (350 mL) (Note 47). The cloudy, white aqueous layer was extracted with ethyl acetate (75 mL x 4) (Note 48) (Figure 12B). The green organic layers were combined in a 500-mL Erlenmeyer flask (Figure 12C), washed with brine (175 mL x 2), dried with anhydrous granular sodium sulfate, and vacuum filtered through a 350 mL fritted funnel with a 24/40 adapter attached to a 1-L rotary evaporation flask. This solution was concentrated via rotary evaporation (300-50 mbar) (Note 49). The residual solvent was removed via high vacuum (30 mbar) to afford diyne monoacid 3 (5.95 g; 93 % yield) (Note 50 and 51) as a viscous dark-brown oil determined to be of 95 % purity via qNMR using 1,3-benzodioxole as internal standard (Note 52) (Figure 12D). The boiling point of diyne monoacid 3 was observed to be 148-150oC at 13 mbar.

Figure 12. Workup of diyne monoacid 3; A. reaction mixture transferred to 1-L separatory funnel; B. extraction with ethyl acetate after acidification with 6 M HCl; C. combined ethyl acetate washes (photos provided by authors); D. diyne monoacid stored in a 20 mL scintillation vial (photo provided by checkers)
C. 4,4-Dimethyl-1,6-heptadiyne (4). An oven-dried, 250-mL, 2-neck round bottom flask (24/40 joints) was equipped with a glass thermometer attached to a glass adapter (24/40), and a rubber septum punctured by an inlet needle attached to a Schlenk line, and an octagonal Teflon-coated stir bar (25 mm x 8 mm). The 2-neck round bottom flask was evacuated and purged with nitrogen in triplicate before diyne monoacid 3 (4.8 g, 28.7 mmol, 1.0 equiv) was added. Acetonitrile (140 mL) was added, followed by CuCl (142 mg, 5 mol%), and the solution was stirred (850 rpm) at ambient temperature (Note 53 and 54) (Figure 13A). Triethylamine (12.2 mL, 88.1 mmol, 3.1 equiv, Note 55) was added via syringe pump (0.75 mL/min) while stirring (850 rpm) at ambient temperature (Note 56) (Figure 13B). The mixture becomes cloudy during the addition of Et3N (Note 57).

Figure 13. General glassware assembly and color changes as triethylamine was added; A. before Et3N addition; B. after Et3N addition (photos provided by checkers)
Upon complete addition of triethylamine, the rubber septum was quickly replaced with a 24/40 reflux condenser, closed with a rubber septum punctured with a needle connected to an argon line, and the reaction mixture was moved to a 70 ℃ pre-heated silicone oil bath (Note 58). The mixture was stirred (850 rpm) and the reaction temperature was kept at 62 ℃ for 2.25 h (Note 59) (Figure 14). Subsequently, the flask was removed from the oil bath, and the mixture was left to stir until it cooled down to room temperature (19-21 ℃ as monitored by the internal thermometer).
Figure 14. The refluxing setup (photo provided by checkers)
The resulting light-brown mixture was transferred to a 500-mL separatory funnel and acidified with 1 M HCl (255 mL) (Figure 15A). The aqueous phase was separated from the brown organic phase, and the aqueous phase was extracted with pentane (60 mL x 4) (Note 60 and 61) (Figure 15B). The combined organic phases were washed with brine (200 mL x 2), dried with anhydrous powdered magnesium sulfate (Note 62), and added to a 500-mL Erlenmeyer flask (Figure 15C).
Figure 15. Glassware used for workup; A. aqueous layer extracted with pentane; B. combined organic phases before washing with brine; C. organic phase dried over magnesium sulfate before purification (photos provided by authors)
Diyne 4 is volatile and care was taken to minimize exposure to reduced pressure. The dried organic layer was purified by gentle vacuum filtration through a small pad of silica gel in a fritted 35-mL funnel with a 14/20 adapter attached to a 14/20-24/40 widening adapter connected to a 1-L rotary evaporation flask (Note 63) (Figure 16A). The organic phase was carefully filtered through the pad of silica gel, and pentane (250 mL) was rapidly passed through the filter without allowing the silica gel to dry out (Note 66). The collected filtrate was carefully evaporated at 0 ℃ (ice bath temperature) using the rotary evaporator (0 rpm; 200 mbar) (Figure 16B). As the pentane evaporated, the pressure was slowly reduced from 200 mbar to 150 mbar. Once only a small amount of solution was left in the 1-L rotary evaporation flask (ca. 25 mL), it was transferred to an 11-dram vial and further concentrated carefully at 150 mbar, thus providing diyne 4 with residual pentane (2.09 g, 57% yield (10 mol% pentane) and 2.74 g (20 mol% pentane), 68% yield) as a clear, colorless liquid of low viscosity (Note 67 and 68). The boiling point of diyne 4 was observed to be 60-61 oC at atmospheric pressure.

Figure 16. Glassware used for purification and rotary evaporator configuration; A. filtration of diyne 4 and pentane through silica gel pad; B. evaporation setup for removing pentane from diyne 4; C. physical appearance of diyne 4 (photos provided by authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with (
dimedone,
acetone,
trifluoromethane sulfonic anhydride,
hydrochloric acid,
diethyl ether,
sodium chloride,
sodium bicarbonate,
sodium sulfate,
ethyl acetate, hexanes,
1,3-benzodioxole,
diisopropylamine,
tetrahydrofuran,
butyllithium,
ammonium chloride,
lithium hydroxide monohydrate,
acetonitrile,
copper(I) chloride,
triethylamine,
pentane,
magnesium sulfate (
Caution: Exercise care in the handling of
trifluoromethanesulfonic anhydride (corrosive),
pyridine (harmful by inhalation),
diisopropylamine (corrosive, toxic, flammable)
butyllithium (flammable, corrosive), and
lithium hydroxide monohydrate (corrosive)).
2. Procedure for the triflation of
dimedone:
Org. Synth. 2024,
101, 124-149 DOI: 10.15227/orgsyn.101.0124 (see reference 6).
3.
Vinylogous acyl triflate (
VAT)
1 exhibited the following spectroscopic characteristics:
1H NMR (400 MHz, CDCl
3) δ 6.07 (t,
J = 1.4 Hz, 1H), 2.55 (d,
J = 1.5 Hz, 2H), 2.32 (s, 2H), 1.14 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ 197.7, 166.2, 123.1, 119.9, 118.1, 116.7, 113.5, 50.4, 42.2, 33.3, 27.8.
19F NMR (376 MHz, CDCl
3) δ -73.87. IR: 2965, 1686, 1648, 1423, 1352, 1206, 1134, 1049, 950, 925, 905, 817, 784, and 754 cm
-1. qNMR purity was determined by making a stock solution containing 45.2 mg of
1,3-benzodioxole (internal standard, 99.5 % purchased from Sigma-Aldrich) and 99.2 mg of VAT measured directly into a small vial. The solution of VAT and internal standard was diluted with 1 mL of CDCl
3 and a 0.1 mL aliquot was transferred to an NMR tube and diluted with 0.6 mL of CDCl
3.
VAT 1 was determined by qNMR to be >97% pure.
4. The authors used a 2-neck round-bottomed flask (14/20 joints) equipped with a 14/20 thermometer adapter.
5. The glass thermometer measured the temperature of the reaction and had a temperature range of -100-50 ℃.
6. The procedure for evacuating and purging glassware with argon in triplicate was repeated for all reactions conducted under inert atmosphere. The authors used nitrogen instead of argon for all reactions requiring inert atmosphere.
7.
Diisopropylamine (99.0 %) was purchased from Sigma-Aldrich and used as received. 1.21 equiv of LDA was made instead of 1.20 equiv.
8.
Tetrahydrofuran (HPLC Grade) was purchased from Fischer Scientific and purified via a Pure Process Technology solvent purification system.
9. The authors used a digital ChemGlass internal thermometer (model CG-3497-100).
10.
Acetone (95%) was purchased from Fischer Chemical and used as received.
11. Once a solution was cooled in a dry ice and
acetone bath, the internal temperature was kept between -68 ℃ and -40 ℃.
12. The temperature of flask B was not monitored and was assumed to be below -40 ℃.
13. 2.5 M
n-butyllithium in hexane (99.0%) was purchased from Sigma-Aldrich and used as received. The authors used 1.6 M
n-butyllithium solution in hexane purchased from Sigma-Aldrich and used as received.
14. The authors used a syringe pump (New Era Pump System Model no. 200).
15. Reagent grade
ethyl acetate (99.5 %) was purchased from Sigma-Aldrich and dried over molecular sieves.
16. When the exact mole ratio of
ethyl acetate to lithium diisopropylamine exceeds 1:1, the resulting ethyl lithioacetate reacts with the excess
ethyl acetate, producing ethyl acetoacetate and negatively impacting the overall yield of product. Thus, a slight excess (e.g., 1.23 equiv) of lithium diisopropylamine is preferable to a slight excess of
ethyl acetate.
17. Pressure in the 1-L round-bottomed flask was partially reduced using the Schlenk line for the cannula addition.
18. TLC plates were purchased from Analytical Chemical (TLC Silica Gel 60, Glass Plates 20 x 20cm, cut into 1.27cm square) and visualized using anisaldehyde stain, which was prepared as follows: To 135 mL of absolute ethanol was added 5 mL of concentrated sulfuric acid and 1.5 mL of glacial acetic acid. The resulting solution was cooled to 0 ℃, and 3.7 mL of
p-anisaldehyde was added. The solution is then stirred vigorously to ensure homogeneity. The solution was stored in a wide mouth 100 mL jar with a plastic cap.
19. The checkers observed a dark-green color (instead of orange) in the second run (see photo below). The color changed to orange upon warming up, and the reaction proceeded in the usual manner.
20.
Ammonium chloride (95%) was purchased from Fischer Scientific and used as received. A half-saturated solution was made by dissolving 100 g of
ammonium chloride in 1-L of distilled water.
21.
Diethyl ether (anhydrous) was purchased from Fischer Chemical and used as received.
22.
Sodium chloride was purchased from Fischer Chemical and diluted with distilled water to make brine.
23.
Sodium sulfate was purchased from Fischer Chemical and used as received.
24.
Ethyl acetate (98%) was purchased from Fischer Chemical and used as received.
25. Hexanes (98%, mixture of isomers) was purchased from Fischer Chemical and used as received.
26. Silica gel was purchased from Fischer Chemical 230-400 micro mesh Grade 60.
27. Sand (20-30 mesh) was purchased from Fischer Chemical and used as received.
28. The authors loaded the sample onto the column in the following manner: the sample was dry loaded neat by adding 20 g of silica to the crude red oil followed by 20 mL of hexanes. This silica/hexanes mixture was concentrated via rotary evaporation to give an orange-red powder which was loaded on top of the silica gel column. An additional 6 g of silica gel and 15 mL of hexanes were added to collect any remaining crude material. The remainder of the crude material (absorbed onto silica gel) was transferred to the column. The 1-L purification column measured 50 mm in diameter and the bulb measured 100 mm in diameter. The checkers loaded the crude mixture onto the column as a solution in hexanes.
29. A 1-L column with 35/20 wide-mouth adapter was packed with 465 g of silica gel and equilibrated with 3 column volumes (~750 mL/1 column volume) of 2%
ethyl acetate/ hexanes. The crude sample was dry loaded, and a thin pad of sample was created by adding 30 mL of hexanes and gently tapping the sides of the column. A layer of sand (258 g) was added on top of this layer of sample and the column bulb was filled with 2%
ethyl acetate/hexanes.
30. Fractions were collected in 25 mm x 50 mm test tubes (purchased from Fischer Scientific). Fractions were collected at 80 mL/min (50 mL/test tube). The checkers collected 125 mL fractions in 125-250 mL Erlenmeyer flasks.
31. The column was run at 2%
ethyl acetate/hexanes for 1 column volume followed by 5%
ethyl acetate/hexanes for 5.5 column volumes. TLCs were taken of all fractions and test tubes 47-94 were collected by filtering through a funnel packed with cotton into a 2-L rotary evaporation flask. The fractions were concentrated via rotary evaporation (300-50 mbar) and residual solvent was removed via high vacuum line (30 mbar) for 0.5 h.
32. The isolated β-keto ester exhibited the following spectroscopic characteristics:
1H NMR
pdf (600 MHz, CDCl
3):
enol form (minor): δ 12.11 (br s, 1H), 5.00 (s, 1H), 4.19 (q,
J = 7.1, 2H), 2.20 (s, 2H), 2.19 (d,
J = 2.6 Hz, 2H), 2.04 (t,
J = 2.6 Hz, 1H), 1.29 (t,
J = 7.1 Hz, 3H), 1.07 (s, 6H);
keto form (major): δ 4.19 (q,
J = 7.1 Hz, 2H), 3.43 (s, 2H), 2.59 (s, 2H), 2.28 (d,
J = 2.6 Hz, 2H), 2.01 (t,
J = 2.6 Hz, 1H), 1.28 (t,
J = 7.1 Hz, 3H), 1.10 (s, 6H). Ratio of keto to enol form = 3.4 : 1 (based on gem-dimethyl integral ratio).
13C NMR
pdf (151 MHz, CDCl
3, k = keto form, e = enol form): δ 202.1 (k), 176.8 (e), 172.8 (e), 167.3 (k), 99.1 (e), 91.8 (k), 82.2 (e), 82.1 (k), 70.7 (k), 61.5 (k), 60.2 (e), 51.7 (k), 51.1 (k), 46.0 (e), 34.3 (e), 33.6 (k), 32.0 (e), 31.3 (k), 27.3 (e), 27.1 (k), 14.4 (e), 14.3 (k). IR (film, ATR): 3290, 2962, 1741, 1713, 1627, 1367, 1311, 1232, 1153, 1032, 807 cm
-1. The purity was determined by qNMR
pdf using 187.6 mg of
1,3-benzodioxole (internal standard)
pdf and 142 mg of β-keto ester
2 (97%). A second run at the same scale provided 8.22 g (71% yield).
33.
Lithium hydroxide monohydrate (98%+) was purchased from Sigma-Aldrich and used as received.
34. The authors used a glass dish filled with ice and water (ice/water bath). The checkers used Julabo FT902 immersion cooler.
35. The internal temperature of increased from 2.7 ℃ to 17.1 ℃ when
lithium hydroxide was added in one portion. The solution was cooled to 3.2 ℃ before proceeding.
36. The clear, upper layer of the 5 M
lithium hydroxide solution was added without disturbing the lower solid layer.
37. After addition of
lithium hydroxide, the temperature of the reaction must be kept between 3.2-5.6 ℃. If the temperature increases above this range, conversion back to β-keto ester will be observed by TLC. If the temperature drastically decreases below this range, the aqueous phase may solidify.
38.
Trifluoromethane sulfonic anhydride (99.5%) was purchased from Oakwood Chemical as used without further purification.
39. Excess
triflic anhydride (3.0 equiv) was added to ensure full triflation of β-keto ester
2.
40. During addition of
triflic anhydride, the internal temperature will increase rapidly. It is important to keep the solution temperature below 5.0 ℃, including by pausing addition as needed.
41. The authors added
sodium chloride to the ice bath during
triflic anhydride addition to aid in cooling the reaction.
Lithium hydroxide monohydrate was added in excess (10 equiv) to eliminate lithium triflate and saponify the diyne monoester to the
diyne monoacid.
42. If added too rapidly, then
lithium hydroxide monohydrate will increase the reaction temperature above 5.6 ℃ prematurely.
43. The aqueous phase was not included in subsequent TLCs because the phases begin to mix after addition of
THF.
44. The layer of solids was collected with the aqueous phase and appeared to diminish as the aqueous phase was repeatedly extracted with
diethyl ether.
45. After transferring the aqueous phase to the separatory funnel, the flask that contained the aqueous phase was rinsed with 10 mL of distilled water between each transfer.
46. The combined organic phase contained β-keto ester and a minor impurity that may be worked-up separately to recover β-keto ester.
47.
Hydrochloric acid (12.1 N) was purchased from Fischer Scientific and diluted with distilled water to make 6 M
HCl.
48. When the aqueous layer is acidified with 6 M
HCl, a small amount of heat will be generated.
49. Upon transferring the material from the 1 L flask to a smaller flask with DCM, the checkers observed formation of a solid crystalline precipitate. The checkers vacuum-filtered the suspension through a celite-packed fritted funnel, and removed the solvent in vacuo. Then, the mixture was once again resuspended in DCM, which caused further precipitation. The solids were again filtered off over celite and the process was repeated two more times, until no significant precipitation was observed. The checkers hypothesize that this precipitate is lithium triflate, extracted into
ethyl acetate layers (
ethyl acetate is known to dissolve lithium triflate) during repeated extractions of the acidified, and highly concentrated aqueous layer. Lithium triflate does not appear to negatively affect the subsequent decarboxylation step (step C).
50. The checkers performed the procedure (step B) twice. First time, the checkers did not attempt to remove the precipitated material described in
Note 49, which resulted in the higher reported yield. Second time, the checkers removed the precipitate as described in the
Note 49, which resulted in the lower reported yield. The qNMR purity refers to the yield obtained from the second attempt (that includes the removal of the precipitate). The second run provided 5.43 g (83% yield) of
3.
51. A small impurity is visible in the β-keto ester NMR spectrum.
52. The isolated
diyne monoacid 3 exhibited the following spectroscopic characteristics:
1H NMR
pdf (600 MHz, CDCl
3) δ: 9.85-7.71 (br s, 1H), 2.40 (s, 2H), 2.21 (d,
J = 2.6 Hz, 2H), 2.03 (t,
J = 2.6 Hz, 1H), 1.11 (s, 6H).
13C NMR
pdf (151 MHz, CDCl
3) δ: 158.1, 90.2, 81.3, 74.7, 71.0, 34.3, 31.3, 31.1, 26.6. IR
pdf (film, ATR): 3299, 2964, 2235, 1683, 1410, 1260, 1070, 901, 782, 755 cm
-1. The purity was determined by qNMR
pdf using 76.9 mg of
1,3-benzodioxole (internal standard) and 100 mg of
diyne monoacid 3 (95%).
53.
Acetonitrile (98%) was purchased from Fischer Chemical and used as received.
54.
Copper(I) chloride (99.99%, trace metal basis) was purchased from Thermo Scientific and used as received.
55.
Triethylamine (99.5%, HPLC grade) was purchased from Thermo Scientific and used as received.
56. The ambient temperature in the lab at the start of the reaction was 21.8 ℃.
57. The color of the reaction mixture is strongly dependent on the purity of
copper(I) chloride used. If CuCl is contaminated with Cu(II) species, the mixture has a darker color (dark red before the addition of
Et3N, and orange-brown after the addition of
Et3N). Checkers did not notice any changes in the reactivity and reaction outcome as a consequence of these observations.
58. The authors used a heating mantle (250 mL) that was purchased from Radleys and used as received. The heating mantle was pre-heated to 70 ℃ and lowered to 65 ℃ once the reaction temperature reached 60 ℃. The checkers used a silicone oil bath heated on a stirring hot plate. The checkers preheated the oil bath and maintained it at [68 ± 1] ℃ throughout the reaction.
59. The checkers observed refluxing in the condenser, which corresponds to the desired product
diyne 4, and not the solvent. The authors did not use a reflux condenser choosing to close the flask with a rubber septum.
60.
Pentane (HPLC grade) was purchased from Fischer Chemical and used as received.
61. The brown organic phase was combined with the
pentane used to extract the aqueous phase and the combined organic phase was kept in an ice bath contained in a small glass dewar.
62.
Magnesium sulfate was purchased from Spectrum Chemical and used as received.
63. Approximately 25 g of silica was used to cover the frit of the vacuum filter and the silica was equilibrated with 35 mL of
pentane.
64. A gentle vacuum was applied via running water.
65. The solution was left in the ice bath for 15 min before reducing pressure to 300 mbar, at which point, it was assumed to be below 5 ℃.
66. As the volume of
pentane in the flask decreased, the pressure was decreased steadily.
67. Simple
diyne 4 exhibited the following spectroscopic characteristics:
1H NMR
pdf (600 MHz, CDCl
3): δ 2.21 (d,
J = 2.6 Hz, 4H), 2.01 (t,
J = 2.7 Hz, 2H), 1.08 (s, 6H).
13C NMR
pdf (151 MHz, CDCl
3): δ 82.0, 70.4, 33.8, 31.0, 26.4. IR
pdf (film, ATR): 3299, 2963, 2118, 1470, 1388, 1262, 1194 cm
-1.
68. The authors measured the purity of
diyne 4 by qNMR using 12.06 mg of
1,3-benzodioxole as the internal standard and 10.51 mg of diyne that was measured directly into an NMR tube placed in a 10-mL graduated cylinder on an analytical balance. The checkers attempted to determine purity by qNMR, however the product
4 evaporated quickly and made the measurement highly inaccurate. The material was assessed to be highly pure by the absence of visible impurities in the
1H and
13C NMR spectra (besides some residual
pentane), and no solids were observed upon evaporation of drops of the product.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
1,6‑Diynes are versatile buildings blocks for organic synthesis and methodology (Figure 17). 1,6‑Heptadiyne, diethyl 2,2-dipropargylmalonate, and other readily available 1,6‑diynes are workhorse substrates for reaction discovery and cyclotrimerization methodology. In contrast, 4,4-dimethyl-1,6-heptadiyne (
4) and its alkyne-substituted derivatives are conspicuously absent from such studies, despite inherent Thorpe-Ingold conformational biases
2 that can facilitate cycloisomerization pathways, and an otherwise-unfunctionalized and relatively inert hydrocarbon framework. The neopentylene tether of
4 also maps conveniently onto numerous polycyclic sesquiterpenes and other targets of interest for synthetic chemistry.
Figure 17. 1,6-Diyne building blocks for organic synthesis and selected natural products bearing gem-dimethylcyclopentane ring fusions
A synthesis of 4,4-dimethyl-1,6-heptadiyne (
4) and its derivatives was previously described by Fleming in 1999 (Figure 18).
3 Two-step Eschenmoser-Tanabe fragmentation of isophorone provides 4,4-dimethyl-6-heptyn-2-one,
4 which can be bis-silylated and then selectively mono-desilylated to provide silylacetylene
5. Dehydration of the ketone is accomplished by LDA-mediated enol phosphorylation and then LDA-mediated elimination of the resulting phosphate, thereby generated diyne monosilane
6. Desilylation then provides
4. Monosilane
6 is a versatile intermediate for making other unsymmetrically substituted neopentylene-tethered (NPT) diynes, e.g., by substitution at the terminal alkyne followed by desilylation.
Figure 18. Prior synthesis 4,4-dimethyl-1,6-heptadiyne
3This new synthesis of
4 compares favorably in terms of step-count and overall yield to the earlier process.
5 Key innovations here include the enolate-mediated fragmentation
6 of
vinylogous acyl triflate 1 to β‑keto ester
2 and the lithium hydroxide-mediated dehydration and saponification of
2 to diyne monocarboxylic acid
3. For example, monoacid
3 was originally prepared from monosilane
6 (e.g., for application in the total synthesis of alcyopterosin E)
7 by carboxylation followed by desilylation, whereas now it is available in three steps and ca. 70% overall yield from dimedone. Copper-catalyzed decarboxylation of
3 provides 4,4-dimethyl-1,6-heptadiyne (
4).
Saponification is slower than dehydration in the conversion of
2 to
3, so saponification optionally can be avoided. Maintaining the reaction temperature at 0 ℃ and limiting the reaction time minimizes saponification,
8 but it can be more convenient and chemoselective to substitute tetrabutylammonium fluoride (TBAF), a milder and less nucleophilic base, for LiOH in the second stage of the process (Figure 19).
Figure 19. Synthesis ethyl 2,7-octadiynoate (diyne monoester 8)
5Multiple options for the synthesis of unsymmetrically substituted neopentylene-tethered (NPT) diynes from β‑keto ester
2 have been developed. As mentioned above, TBAF-mediated triflate elimination provides diyne monoester
8. Either ester
8 or acid
3 can be methylated or silylated on the terminal alkyne, and alkynoic acids can be decarboxylated as desired. Ester
8 is a good substrate for Sonogashira couplings (Figure 19), and subsequent saponification and decarboxylative Sonogashira coupling can produce unsymmetrically diarylated diynes.
5Figure 20. Alkylation and Zard fragmentation as an alternative approach to differentially substituted 4,4-dimethyl-1,6-heptadiyne derivatives
α-Alkylated derivatives of βketo‑ ester
2 are good substrates for Zard fragmentation
9 as a complementary approach to mono- or dialkylated diynes (Figure 20). The high yielding and scalable synthesis of βketo‑ ester
2 and multiple options for dehydration and functionalization thus offer versatile and convenient access to diverse NPT diynes for synthesis and methodology.
Figure 21. Synthesis of neopentylene-tethered 1,6 diynes from dimedone
In summary, a high-yielding, three-step process can be used to convert dimedone into 5,5-dimethyl-2,7-octadiynoates for further functionalization (Figure 21). Copper-catalyzed decarboxylation gives 4,4-dimethyl-1,6-heptadiyne (4). Alternatively, alkylation and/or arylation provides diverse neopentylene-tethered 1,6‑diynes as versatile substrates for annulation methodology and/or as building blocks for target-oriented synthesis.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Ethyl 5,5-Dimethyl-3-oxooct-7-ynoate: 7-Octynoic acid, 5,5-dimethyl-3-oxo-, ethyl ester; (2) (2557181-86-7)
5,5-Dimethyl-2,7-octadiynoic acid: 5,5-Dimethyl-2,7-octadiynoic acid; (3) (500596-39-4)
4,4-Dimethyl-1,6-heptadiyne; (4) (148181-34-4)

|
Amir Tavakoli obtained his BS degree in chemistry from Sharif University of Technology in Tehran. He then went on to pursue his graduate studies in synthetic organic chemistry in Prof. Greg Dudley's lab at West Virginia University in Morgantown and earned his PhD in 2023. He is currently a process chemist at Vertex Pharmaceuticals. |

|
Nathaniel Selvaraj received his B.Sc. in Chemistry at West Virginia University in May 2022. He is currently pursuing his Ph.D. at West Virginia University under the supervision of Brian Popp and Gregory Dudley. He is currently investigating the mechanism of transition metal-catalyzed cyclotrimerizations with tethered 1,6-diynes. |

|
Gregory B. Dudley received a B.A. from FSU in 1995 and a Ph.D. in 2000 under the direction of Rick Danheiser at MIT, then completed an NIH Postdoctoral Fellowship with Samuel Danishefsky at the Sloan-Kettering Institute for Cancer Research. He started his independent career at FSU in 2002 and moved to WVU in 2016. The fragmentation / olefination methodology featured herein has figured prominently in his research program over the past two decades. In Fall 2025, he moved to the University of Houston to serve as Dean of Natural Sciences and Mathematics. |

|
Aleksa Milosavljevic received a B.Sc. in Chemistry in 2019 at the University of Belgrade in Serbia and a Ph.D. in 2024 in the group of Prof. Alison J. Frontier at the University of Rochester. During his Ph.D., he investigated nitrogen-interrupted halo-Nazarov cascades, and completed a total synthesis of an indole diterpenoid natural product tubingensin A. He is currently a postdoctoral researcher in the group of Prof. Dirk Trauner at the University of Pennsylvania. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved